
Large Phenotypic Variation of Individuals from a Family with a Novel ASPM Mutation Associated with Microcephaly, Epilepsy, and Behavioral and Cognitive Deficits

 , , , , , and
, , , , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Neuropsychological Rating
2.3. Whole Exome Sequencing
2.4. ASPM Expression Analysis
2.5. MRI Processing and Analysis
3. Results
3.1. Case A (II.2)
3.2. Case B (II.3)
3.3. Case C (II.4)
3.4. Cognitive and Disability Assessment of the Individual Patients
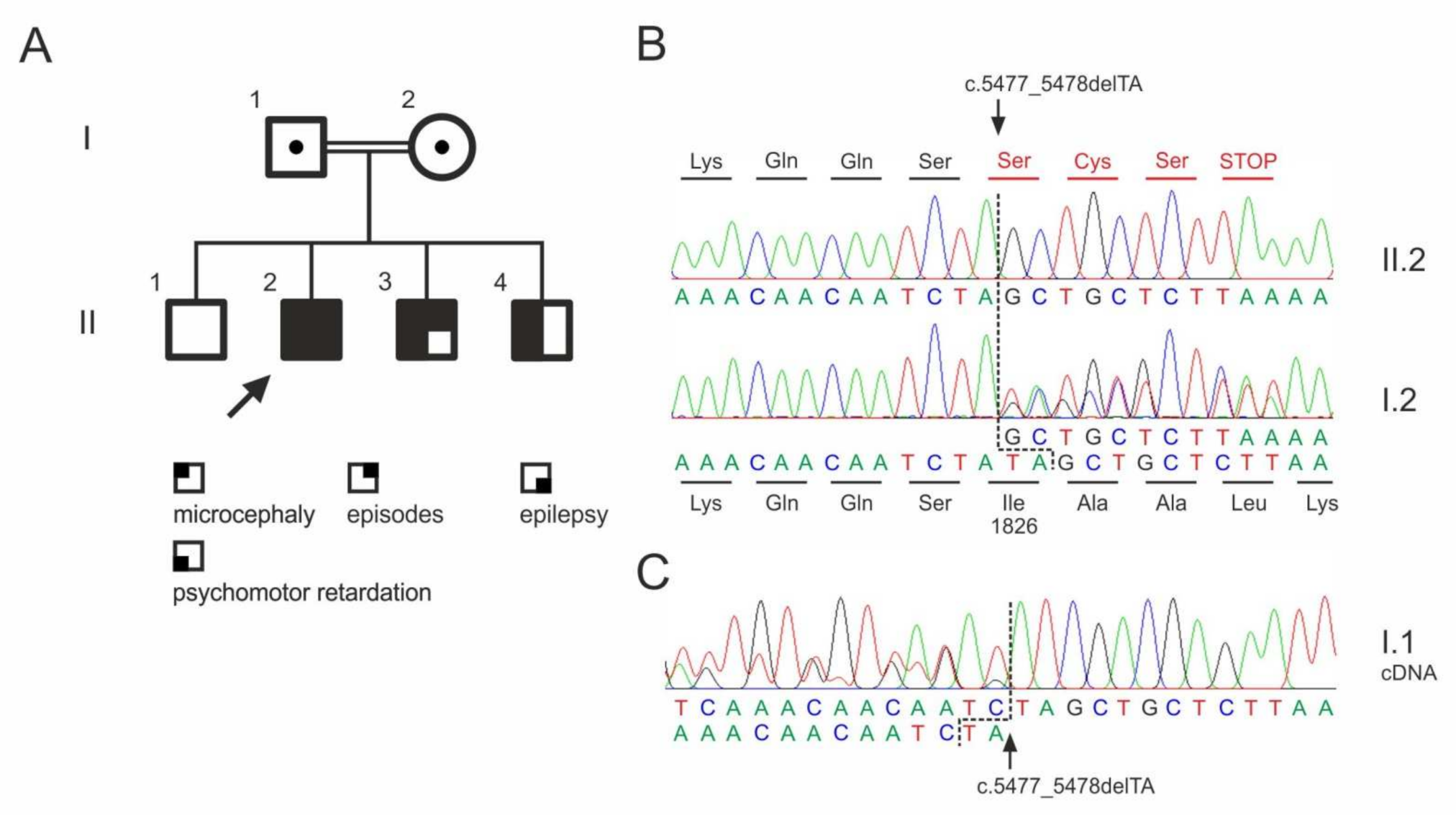
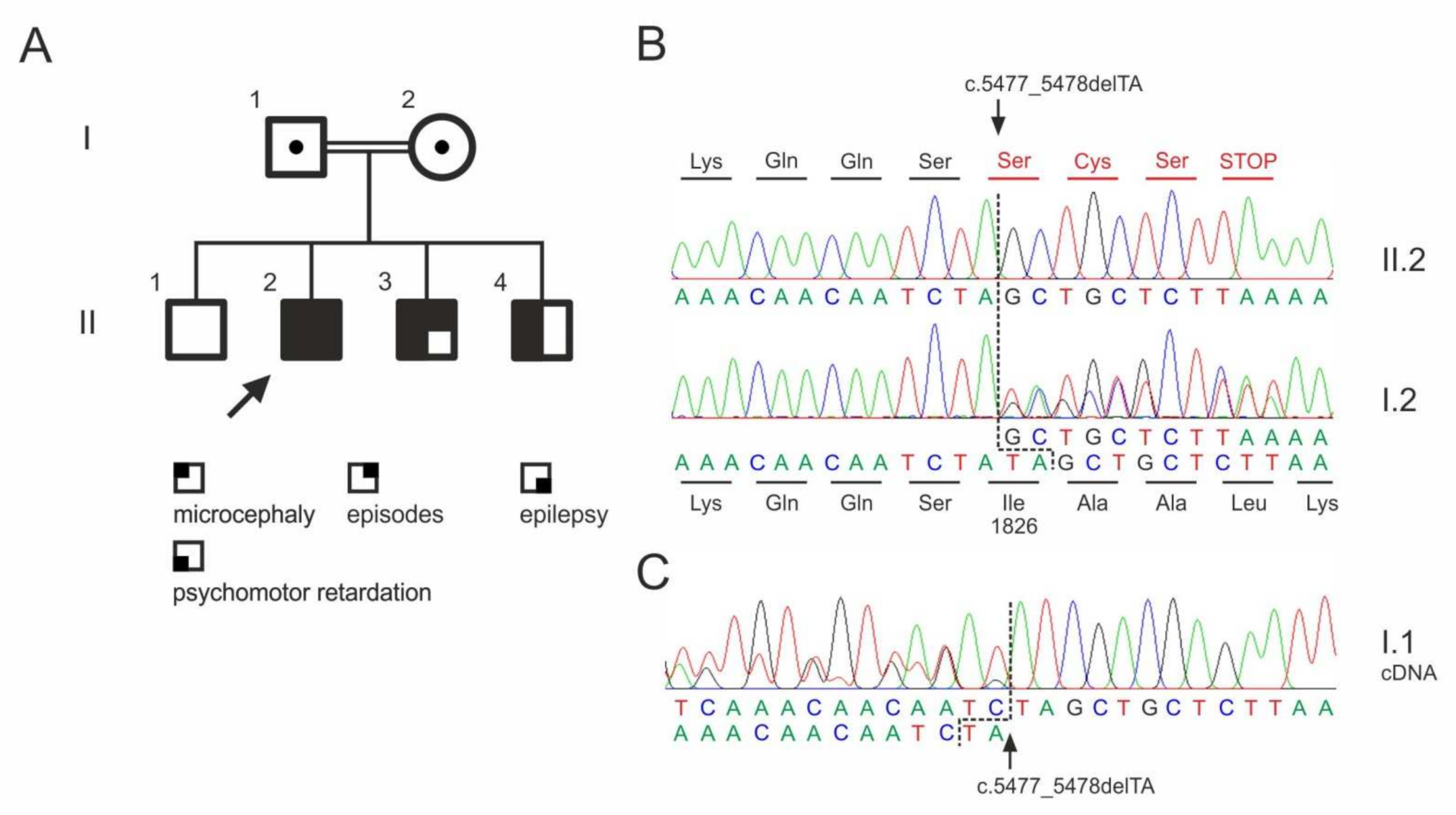
3.5. Whole Exome Sequencing
3.6. MRI Processing and Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Weber, Y.G.; Biskup, S.; Helbig, K.L.; Von Spiczak, S.; Lerche, H. The role of genetic testing in epilepsy diagnosis and management. Expert Rev. Mol. Diagn. 2017, 17, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Møller, R.S.; Hammer, T.B.; Rubboli, G.; Lemke, J.R.; Johannesen, K.M. From next-generation sequencing to targeted treatment of non-acquired epilepsies. Expert Rev. Mol. Diagn. 2019, 19, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Zaqout, S.; Morris-Rosendahl, D.; Kaindl, A.M. Autosomal Recessive Primary Microcephaly (MCPH): An Update. Neuropediatrics 2017, 48, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Woods, C.G.; Bond, J.; Enard, W. Autosomal recessive primary microcephaly (MCPH): A review of clinical, molecular, and evolutionary findings. Am. J. Hum. Genet. 2005, 76, 717–728. [Google Scholar] [CrossRef] [Green Version]
- Létard, P.; Drunat, S.; Vial, Y.; Duerinckx, S.; Ernault, A.; Amram, D.; Arpin, S.; Bertoli, M.; Busa, T.; Ceulemans, B.; et al. Autosomal recessive primary microcephaly due to ASPM mutations: An update. Hum. Mutat. 2018, 39, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Passemard, S.; Titomanlio, L.; Elmaleh, M.; Afenjar, A.; Alessandri, J.-L.; Andria, G.; de Villemeur, T.B.; Boespflug-Tanguy, O.; Burglen, L.; Del Giudice, E.; et al. Expanding the clinical and neuroradiologic phenotype of primary microcephaly due to ASPM mutations. Neurology 2009, 73, 962–969. [Google Scholar] [CrossRef]
- Abdel-Hamid, M.S.; Ismail, M.F.; Darwish, H.A.; Effat, L.K.; Zaki, M.S.; Abdel-Salam, G.M.H. Molecular and phenotypic spectrum of ASPM-related primary microcephaly: Identification of eight novel mutations. Am. J. Med. Genet. A 2016, 170, 2133–2140. [Google Scholar] [CrossRef] [PubMed]
- Raven, J.; Court, J.H. Manual for Raven’s Progressive Matrices and Vocabulary Scales. In Research Supplement No. 4: Additional National and American Norms, and Summaries of Normative, Reliability, and Validity Studies; Oxford Psychologists Press: Oxford, UK, 1989. [Google Scholar]
- Kaufman, A.S.; Kaufman, N.L. Kaufman Assessment Battery for Children, 2nd ed.; American Guidance Service: Circle Pines, MN, USA, 2004. [Google Scholar]
- Chan, C.J.; Zou, G.; Wiebe, S.; Speechley, K.N. Global assessment of the severity of epilepsy (GASE) scale in children: Validity, reliability, responsiveness. Epilepsia 2015, 56, 1950–1956. [Google Scholar] [CrossRef]
- Funke, U.-N.; Schüwer, U.; Themann, P.; Gerdes, N. Selbständigkeits- Index für die Neurologische und Geriatrische Rehabilitation SINGER: Manual zur Stufenzuordnung. 2., Vollständig Überarbeitete und Erweitertete Auflage; S. Roderer Verlag: Regensburg, Germany, 2018. [Google Scholar]
- Berzina, G.; Sveen, U.; Paanalahti, M.; Sunnerhagen, K.S. Analyzing the modified Rankin Scale using concepts of the international classification of functioning, disability and health. Eur. J. Phys. Rehabil. Med. 2016, 52, 203–213. [Google Scholar]
- Basmanav, F.B.; Oprisoreanu, A.-M.; Pasternack, S.M.; Thiele, H.; Fritz, G.; Wenzel, J.; Größer, L.; Wehner, M.; Wolf, S.; Fagerberg, C.; et al. Mutations in POGLUT1, encoding protein O-glucosyltransferase 1, cause autosomal-dominant Dowling-Degos disease. Am. J. Hum. Genet. 2014, 94, 135–143. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Huppertz, H.-J.; Kröll-Seger, J.; Klöppel, S.; Ganz, R.E.; Kassubek, J. Intra- and interscanner variability of automated voxel-based volumetry based on a 3D probabilistic atlas of human cerebral structures. Neuroimage 2010, 49, 2216–2224. [Google Scholar] [CrossRef] [PubMed]
- Huppertz, H.-J.; Möller, L.; Südmeyer, M.; Hilker, R.; Hattingen, E.; Egger, K.; Amtage, F.; Respondek, G.; Stamelou, M.; Schnitzler, A.; et al. Differentiation of neurodegenerative parkinsonian syndromes by volumetric magnetic resonance imaging analysis and support vector machine classification. Mov. Disord. 2016, 31, 1506–1517. [Google Scholar] [CrossRef]
- Dale, A.M.; Fischl, B.; Sereno, M.I. Cortical surface-based analysis. I. Segmentation and surface reconstruction. Neuroimage 1999, 9, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rentzsch, P.; Schubach, M.; Shendure, J.; Kircher, M. CADD-Splice—Improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 2021, 13, 31. [Google Scholar] [CrossRef]
- Johnson, M.B.; Sun, X.; Kodani, A.; Borges-Monroy, R.; Girskis, K.M.; Ryu, S.C.; Wang, P.P.; Patel, K.; Gonzalez, D.M.; Woo, Y.M.; et al. Aspm knockout ferret reveals an evolutionary mechanism governing cerebral cortical size. Nature 2018, 556, 370–375. [Google Scholar] [CrossRef]
- Ensembl Genome Browser. Available online: https://www.ensembl.org/Homo_sapiens/Transcript/ProteinSummary?db=core;g=ENSG00000066279;r=1:197084121-197146694;t=ENST00000367409 (accessed on 21 December 2021).
- Bond, J.; Roberts, E.; Mochida, G.H.; Hampshire, D.J.; Scott, S.; Askham, J.M.; Springell, K.; Mahadevan, M.; Crow, Y.J.; Markham, A.F.; et al. ASPM is a major determinant of cerebral cortical size. Nat Genet. 2002, 32, 316–320. [Google Scholar] [CrossRef]
- Mekel-Bobrov, N.; Gilbert, S.L.; Evans, P.D.; Vallender, E.J.; Anderson, J.R.; Hudson, R.R.; Tishkoff, S.A.; Lahn, B.T. Ongoing adaptive evolution of ASPM, a brain size determinant in Homo sapiens. Science 2005, 309, 1720–1722. [Google Scholar] [CrossRef]
- Kouprina, N.; Pavlicek, A.; Collins, N.K.; Nakano, M.; Noskov, V.N.; Ohzeki, J.; Mochida, G.H.; Risinger, J.I.; Goldsmith, P.; Gunsior, M.; et al. The microcephaly ASPM gene is expressed in proliferating tissues and encodes for a mitotic spindle protein. Hum. Mol. Genet. 2005, 14, 2155–2165. [Google Scholar] [CrossRef]
- OMIM. Available online: https://www.omim.org/entry/605481?search=ASPM&highlight=aspm (accessed on 21 December 2021).
- Rahit, K.M.; Tarailo-Graovac, M. Genetic modifiers and rare Mendelian disease. Genes 2020, 11, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Patient | II.2 | II.3 | II.4 |
|---|---|---|---|
| RPM * | <55 | <55 | <55 |
| KABC-II ** | 45 | 48 | 48 |
| GASE *** | 5 | n.a. | n.a. |
| mRS **** | 3 | 2 | 3 |
| Barthel index | 100 | 100 | 100 |
| SINGER physical activities (%) ***** | 100 | 100 | 100 |
| SINGER cognition (%) ***** | 45 | 55 | 48 |
| SINGER household (%) ***** | 20 | 60 | 40 |
| SINGER total (%) ***** | 74 | 80 | 76 |
| Gene Name | Genomic Position (GRCh 38) | Amino Acid Change | Allele Frequency # | Homozygous/Hemizygous # |
|---|---|---|---|---|
| APP | 21:26000158G > A | p.Thr297Met | 52/282,588 | 2 |
| TRIM67 | 1:231206722G > A | p.Arg584Gln | 144/277,698 | 1 |
| ASPM | 1:197103775_197103776del | p.Ile1826Serfs * 4 | n.a. | n.a. |
| LUZP4 | X:115289767C > G | p.Ser3Trp | 160/204,902 | 2 / 92 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
von Wrede, R.; Schidlowski, M.; Huppertz, H.-J.; Rüber, T.; Ivo, A.; Baumgartner, T.; Hallmann, K.; Zsurka, G.; Helmstaedter, C.; Surges, R.; et al. Large Phenotypic Variation of Individuals from a Family with a Novel ASPM Mutation Associated with Microcephaly, Epilepsy, and Behavioral and Cognitive Deficits. Genes 2022, 13, 429. https://doi.org/10.3390/genes13030429
von Wrede R, Schidlowski M, Huppertz H-J, Rüber T, Ivo A, Baumgartner T, Hallmann K, Zsurka G, Helmstaedter C, Surges R, et al. Large Phenotypic Variation of Individuals from a Family with a Novel ASPM Mutation Associated with Microcephaly, Epilepsy, and Behavioral and Cognitive Deficits. Genes. 2022; 13(3):429. https://doi.org/10.3390/genes13030429
Chicago/Turabian Stylevon Wrede, Randi, Martin Schidlowski, Hans-Jürgen Huppertz, Theodor Rüber, Anja Ivo, Tobias Baumgartner, Kerstin Hallmann, Gábor Zsurka, Christoph Helmstaedter, Rainer Surges, and et al. 2022. "Large Phenotypic Variation of Individuals from a Family with a Novel ASPM Mutation Associated with Microcephaly, Epilepsy, and Behavioral and Cognitive Deficits" Genes 13, no. 3: 429. https://doi.org/10.3390/genes13030429