
Identification of Key Modules and Genes Associated with Major Depressive Disorder in Adolescents

Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Methods
2.2.1. RNA-Seq Data Generation and Processing
2.2.2. Weighted Co-Expression Network Construction
2.2.3. Module–Trait Correlation and Key Module Identification
2.2.4. Functional Enrichment Analysis of Genes in Key Modules
2.2.5. Hub Genes Identification
3. Results
3.1. RNA-Seq Data and Analysis
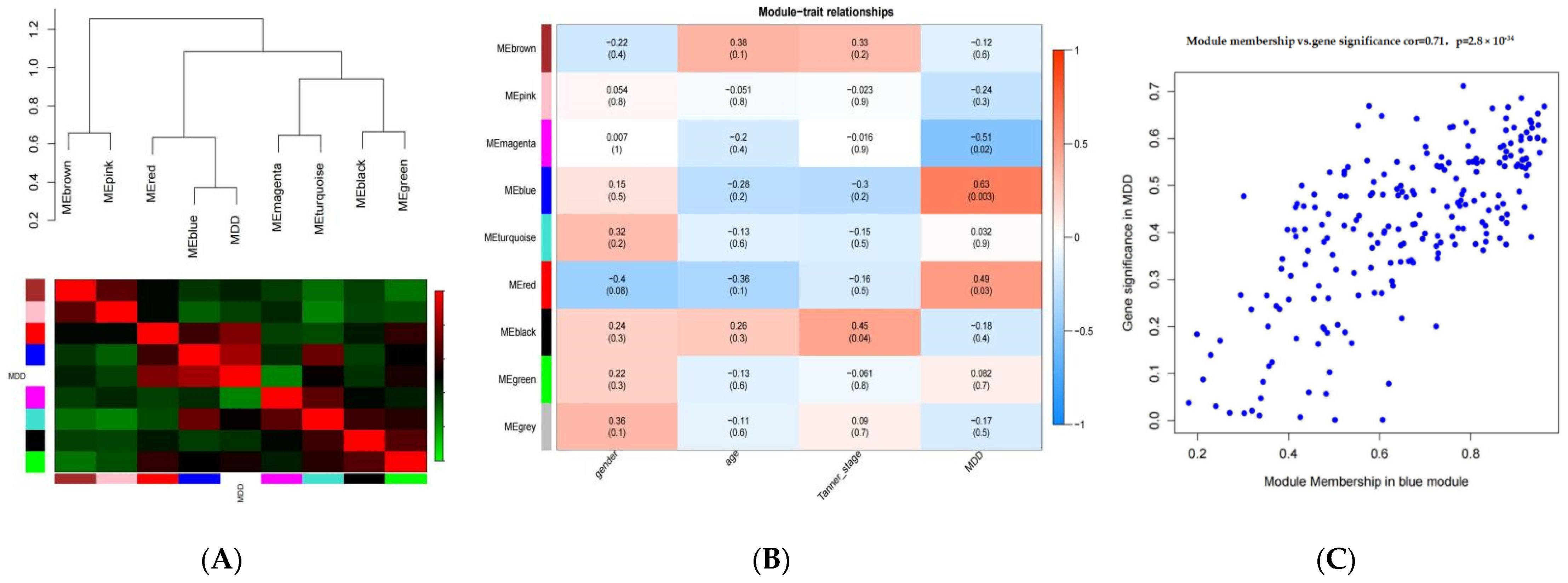
3.2. Identification of Key Modules
3.3. Identification of Key Co-Expression Network Modules for MDD
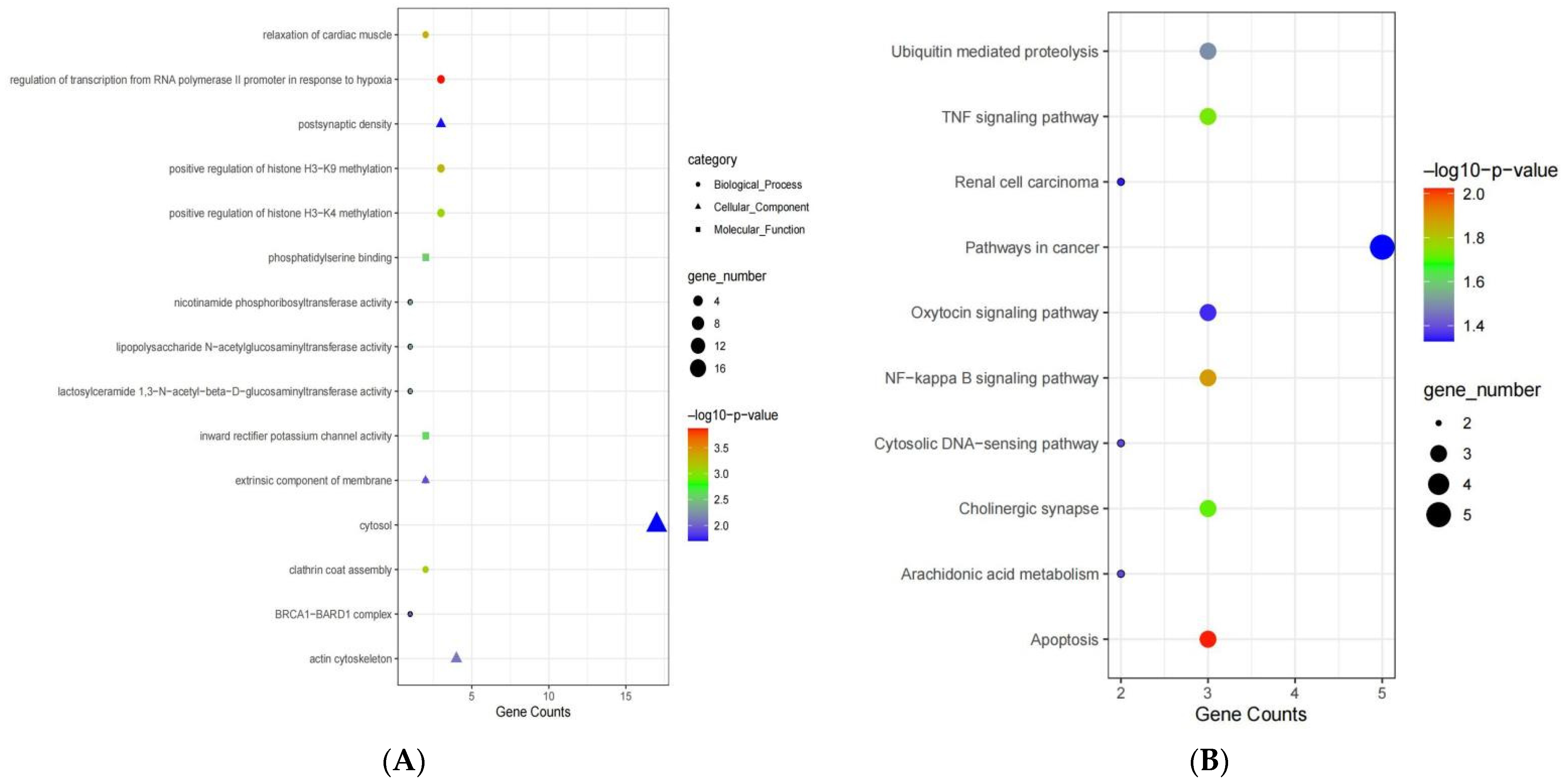
3.4. Functional Enrichment Analysis of Genes in the Blue Module
3.5. Gene Interactions within Blue Module and Hub Genes Identification
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferrari, A.J.; Charlson, F.J.; Norman, R.E.; Patten, S.B.; Freedman, G.; Murray, C.J. Burden of depressive disorders by country, sex, age, and year: Findings from the global burden of disease study. PLoS Med. 2010, 10, e1001547. [Google Scholar]
- Whiteford, H.A.; Degenhardt, L.; Rehm, J.; Baxter, A.J.; Ferrari, A.J.; Erskine, H.E.; Vos, T. Global burden of disease attributable to mental and substance use disorders: Findings from the global burden of disease dtudy. Lancet 2010, 382, 1575–1586. [Google Scholar] [CrossRef]
- Cole, D.A.; Tram, J.M.; Martin, J.M.; Hoffman, K.B.; Ruiz, M.D.; Jacquez, F.M.; Maschman, T.L. Individual differences in the emergence of depressive symptoms in children and adolescents: A longitudinal investigation of parent and child reports. J. Abnorm. Psychol. 2002, 111, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Rice, F.; Riglin, L.; Lomax, T.; Souter, E.; Potter, R.; Smith, D.J.; Thapar, A. Adolescent and adult differences in major depression symptom profiles. J. Affect. Disord. 2018, 243, 175–181. [Google Scholar] [CrossRef] [Green Version]
- Lakdawalla, Z.; Hankin, B.L.; Mermelstein, R. Cognitive theories of depression in children and adolescents: A conceptual and quantitative review. Clin. Child. Fam. Psychol. Rev. 2007, 10, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, H.; Gong, Y.; Ungar, M. Stressful events and depression among Chinese adolescents: The mitigating role of protective factors. Sch. Psychol. Int. 2013, 34, 501. [Google Scholar] [CrossRef]
- Lewinsohn, P.M.; Rohde, P.; Seeley, J.R.; Klein, D.N.; Gotlib, I.H. Psychosocial functioning of young adults who have experienced and recovered from major depressive disorder during adolescence. J. Abnorm. Psychol. 2003, 112, 353–363. [Google Scholar] [CrossRef]
- Merikangas, K.R.; He, J.P.; Burstein, M.; Swanson, S.A.; Avenevoli, S.; Cui, L. Lifetime prevalence of mental disorders in U.S. adolescents: Results from the national comorbidity survey replication–adolescent supplement. Adolesc. Psychiatry 2010, 49, 980–989. [Google Scholar] [CrossRef] [Green Version]
- Birmaher, B.; Dahl, R.E.; Perel, J.; Williamson, D.E.; Nelson, B.; Stull, S. Corticotropin-releasing hormone challenge in prepubertal major depression. Biol. Psychiatry 1996, 39, 267–277. [Google Scholar] [CrossRef]
- Ghaziuddin, N.; King, C.A.; Weich, K.B. Serotonin dusregulation in adolescents with major depression: Hormone response to meta-chlorophenylpiperazine infusion. Psychiatry Res. 2000, 95, 83–194. [Google Scholar] [CrossRef]
- Malkesman, O.; Braw, Y.; Ram, E.; Maayan, R.; Weizman, A.; Kinor, N. Dehydroepiandrosterone and monoamines in the limbic system of a genetic animal model of childhood depression. Eur. Neuropsychopharmacol. 2008, 18, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Kessler, R.C.; Avenevoli, S.; Merikangas, K.R. Mood disorders in children and adolescents: An epidemiologic perspective. Biol. Psychiatry 2001, 49, 1002–1014. [Google Scholar] [CrossRef]
- Hasler, G.; Pine, D.S.; Kleinbaum, D.G.; Gamma, A.; Luckenbaugh, D.; Ajdacic, V.; Angst, J. Depressive symptoms during childhood and adult obesity. Zur. Cohort. Study Mol. Psychiatry 2005, 10, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Aronen, E.T.; Soininen, M. Childhood depressive symptoms predict psychiatric problems in young adults. Can. J. Psychiatry 2000, 45, 465–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynder, E.L.; Graham, E.A. Tobacco smoking as a possible etiologic factor in bronchiogenic carcinoma. Bull. WHO 2005, 83, 146–153. [Google Scholar]
- Fan, H.; Sun, X.; Guo, W. Differential expression of microRNA in peripheral blood mononuclear cells as specific biomarker for major depressive disorder patients. J. Psychiatr. 2014, 59, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, F.N.; Costa, A.P.; Ghisleni, G.; Diaz, A.P.; Rodrigues, A.L.S.; Peluffo, H.; Kaster, M.P. NLRP3 inflammasome-driven pathways in depression. Clin. Preclin. Find. 2017, 64, 367–383. [Google Scholar]
- Redei, E.E.; Andrus, B.M.; Kwasny, M.J.; Seok, J.; Cai, X.; Ho, J. Blood transcriptomic biomarkers in adult primary care patients with major depressive disorder undergoing cognitive behavioral therapy. Transl. Psychiatry 2014, 4, e442. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Jiang, Y.; Zhong, X.; Cox, N.J.; Liu, C.; Gamazon, E.R. A unified framework for joint-tissue transcriptome-wide association and Mendelian randomization analysis. Nat. Genet. 2020, 52, 1239–1246. [Google Scholar] [CrossRef]
- Gerring, Z.F.; Gamazon, E.R.; Derks, E.M. A gene co-expression network-based analysis of multiple brain tissues reveals novel genes and molecular pathways underlying major depression. PLoS Genet. 2019, 15, e1008245. [Google Scholar] [CrossRef] [Green Version]
- Jansen, R.; Penninx, B.; Madar, V. Gene expression in major depressive disorder. Mol. Psychiatry 2015, 21, 339. [Google Scholar] [CrossRef] [PubMed]
- Levinson, D.F.; Mostafavi, S.; Milaneschi, Y.; Rivera, M.; Ripke, S.; Wray, N.R. Genetic studies of major depressive disorder: Why are there no genome-wide association study findings and what can we do about it? Biol. Psychiatry 2014, 76, 510–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belzeaux, R.; Bergon, A.; Jeanjean, V.; Loriod, B.; Formisano-Tréziny, C.; Verrier, L. Responder and nonresponder patients exhibit different peripheral transcriptional signatures during major depressive episode. Transl. Psychiatry 2012, 2, e185. [Google Scholar] [CrossRef] [PubMed]
- Segman, R.H.; Goltser-Dubner, T.; Weiner, I.; Canetti, L.; Hochner-Celnikier, D. Blood mononuclear cell gene expression signature of postpartum depression. Mol. Psychiatry 2009, 15, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.I.; Lim, S.W.; Myung, W.; Kim, D.K.; Lee, S.Y. Differentially expressed genes related to major depressive disorder and antidepressant response: Genome-wide gene expression analysis. Exp. Mol. Med. 2018, 50, 92. [Google Scholar] [CrossRef] [Green Version]
- Yi, Z.; Li, Z.; Yu, S.; Yuan, C.; Hong, W. Blood-based gene expression profiles models for classification of subsyndromal symptomatic depression and major depressive disorder. PLoS ONE 2012, 7, e31283. [Google Scholar] [CrossRef]
- Zhu, Y.; Strachan, E.; Fowler, E.; Bacus, T.; Roy-Byrne, P.; Zhao, J. Genome-wide profiling of DNA methylome and transcriptome in peripheral blood monocytes for major depression: A monozygotic discordant twin study. Transl. Psychiatry 2019, 9, 215. [Google Scholar] [CrossRef] [Green Version]
- Hiroaki, H.; Seiji, N.; Fuyuko, Y. Integrated profiling of phenotype and blood transcriptome for stress vulnerability and depression. J. Psychiatr. Res. 2018, 104, 202–210. [Google Scholar]
- McKinney, B.A.; White, B.C.; Grill, D.E.; Li, P.W.; Kennedy, R.B.; Poland, G.A. A gene-wise adaptive-K nearest-neighbor feature selection tool for finding gene-gene interactions and main effects in mRNA-Seq gene expression data. PLoS ONE. 2013, 8, e81527. [Google Scholar] [CrossRef]
- Girvan, M.; Newman, E.J. Community structure in social and biological networks. Proc. Natl. Acad. Sci. USA 2002, 99, 7821–7826. [Google Scholar] [CrossRef] [Green Version]
- Oldham, M.C.; Horvath, S.; Geschwind, D.H. Conservation and evolution of gene coexpression networks in human and chimpanzee brains. Proc. Natl. Acad. Sci. USA 2006, 3, 17973–17978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, S.; Bhattacharyya, D.K.; Kalita, J.K. Reconstruction of gene co-expression network from microarray data using local expression patterns. BMC Bioinform. 2014, 15, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, T.T.; Jonathan, S.; Hideo, S.; Masaya, M.; Kent, T.T.; White, B.C. Identification and replication of rna-seq gene network modules associated with depression severity. Transl. Psychiatry 2018, 8, 180. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Cheng, L. Two gene co-expression modules differentiate psychotics and controls. Mol. Psychiatry. 2013, 18, 1308–1314. [Google Scholar] [CrossRef] [Green Version]
- Jong, S.A. Gene co-expression network in whole blood of schizophrenia patients is independent of antipsychotic-use and enriched for brainexpressed genes. PLoS ONE 2012, 7, e39498. [Google Scholar]
- Torkamani, A.; Dean, B.; Schork, N.J.; Thomas, E.A. Coexpression network analysis of neural tissue reveals perturbations in developmental processes in schizophrenia. Genome Res. 2010, 20, 403–412. [Google Scholar] [CrossRef] [Green Version]
- Voineagu, I. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011, 474, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.C. A conserved BDNF, glutamate and GABA-enriched gene module related to human depression identified by coexpression metaanalysis and DNA variant genome-wide association studies. PLoS ONE 2014, 9, e90980. [Google Scholar]
- Gaiteri, C.; Ding, Y.; French, B.; Tseng, G.C.; Sibille, E. Beyond modules and hubs: The potential of gene coexpression networks for investigating molecular mechanisms of complex brain disorders. Genes Brain Behave 2014, 13, 13–24. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 124. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.F. Parallel algorithms for hierarchical clustering. Parallel Comput. 1995, 21, 1313–1325. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Zhang, B.; Horvath, S. Defining clusters from a hierarchical cluster tree: The Dynamic Tree Cut package for R. Bioinformatics 2008, 24, 719–720. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Horvath, S. Understanding network concepts in modules. BMC Syst. Biol. 2007, 1, 24. [Google Scholar] [CrossRef] [Green Version]
- Qi, G.; Kong, W.; Mou, X.; Wang, S. A new method for excavating feature lncrna in lung adenocarcinoma based on pathway crosstalk analysis. J. Cell. Biochem. 2018, 12, 1–13. [Google Scholar] [CrossRef]
- Rhead, B.; Shao, X.; Quach, H.; Ghai, P.; Barcellos, L.F.; Bowcock, A.M. Global expression and CpG methylation analysis of primary endothelial cells before and after TNFa stimulation reveals gene modules enriched in inflammatory and infectious diseases and associated DMRs. PLoS ONE 2020, 15, e0230884. [Google Scholar]
- Zhao, Y.; Wang, L.; Wu, Y.J.; Lu, Z.Q.; Zhang, S.Y. Genome-wide study of key genes and scoring system as potential noninvasive biomarkers for detection of suicide behavior in major depression disorder. Bioengineered 2020, 11, 1189–1196. [Google Scholar] [CrossRef]
- Gerring, Z.F. Dissecting genetically regulated gene expression in major depression. Biol. Psychiatry 2021, 89, 6. [Google Scholar] [CrossRef]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F. Systematic RNA interference reveals that oncogenic kras-driven cancers require tbk1. Nature 2009, 462, 108–112. [Google Scholar] [CrossRef]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, M.A. Rating scale for depression. J. Neurol. Neurosurg. Psychiatry 1960, 23, 56–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders V(DSM-V); American Psychiatric Association: Washington, DC, USA, 2013; pp. 83–282. [Google Scholar]
- Taylor, S.J.; Whincup, P.H.; Hindmarsh, P.C.; Lampe, F.; Odoki, K.; Cook, D.G. Performance of a new pubertal self-assessment questionnaire: A preliminary study. Paediatr. Perinat. Epidemiol. 2001, 15, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Boutros, P.C.; Okey, A.B. Unsupervised pattern recognition: An introduction to the whys and wherefores of clustering microarray data. Brief. Bioinform. 2005, 4, 316–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, K.; Bing, Z.; Cao, G.; Guo, L.; Cao, Y.; Jiang, H.; Zhang, M. Identify the signature genes for diagnose of uveal melanoma by weight gene co-expression network analysis. Int. J. Ophthalmol. 2015, 2, 269–274. [Google Scholar]
- van Dam, S.; Cordeiro, R.; Craig, T.; van Dam, J.; Wood, S.H.; de Magalhães, J.P. Gene Friends: An online co-expression analysis tool to identify novel gene targets for aging and complex diseases. BMC Genom. 2012, 13, 535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Langfelder, P.; Horvath, S. Comparison of co-expression measures: Mutual information, correlation, and model based indices. BMC Bioinform. 2012, 13, 328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rau, C.; Wisniewski, N.; Orozco, L.D.; Bennett, B.J.; Weiss, J.N.; Lusis, A.J. Maximal information component analysis: A novel non-linear network analysis method. Front. Genet. 2013, 4, 28. [Google Scholar] [CrossRef] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, 14. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef]
- Benjamini, Y.; Drai, D.; Elmer, G.; Kafkafi, N.; Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 2001, 125, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Y.; Cai, J.H.; Tsai, J.J.; Wang, C.C. Identification of Hub Genes Associated with Development of Head and Neck Squamous Cell Carcinoma by Integrated Bioinformatics Analysis. Front. Oncol. 2020, 10, 681. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, Y.; Zhang, Z.; Li, H. Identification of KIF18B as a hub candidate gene in the metastasis of clear cell renal cell carcinoma by weighted gene co-expression network analysis. Front. Genet. 2020, 11, 905. [Google Scholar] [CrossRef]
- Li, Z.; Chyr, J.; Jia, Z.; Wang, L.; Hu, X.; Wu, X.; Song, C. Identification of hub genes associated with hypertension and their interaction with miRNA based on weighted gene coexpression network analysis (WGCNA) analysis. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2020, 26, e923514. [Google Scholar] [CrossRef]
- Niemira, M.; Collin, F.; Szalkowska, A.; Bielska, A.; Chwialkowska, K.; Reszec, J.; Niklinski, J.; Kwasniewski, M.; Kretowski, A. Molecular signature of subtypes of non-small-cell lung cancer by large-scale transcriptional profiling: Identification of key modules and genes by weighted gene co-expression network analysis (WGCNA). Cancers 2019, 12, 37. [Google Scholar] [CrossRef] [Green Version]
- Gaujoux, R.; Seoighe, C. CellMix: A comprehensive toolbox for gene expression deconvolution. Bioinformation 2013, 29, 2211. [Google Scholar] [CrossRef]
- Heim, C.; Newport, D.J.; Mletzko, T.; Miller, A.H.; Nemeroff, C.B. The link between childhood trauma and depression: Insights from hpa axis studies in humans. Psychoneuroendocrinology 2008, 33, 693–710. [Google Scholar] [CrossRef]
- Slavich, G.M.; Irwin, M.R. From stress to inflammation and major depressive disorder: A social signal transduction theory of depression. Psychol. Bull. 2014, 140, 774–815. [Google Scholar] [CrossRef]
- Park, C.; Rosenblat, J.; Brietzke, E. Stress, Epigenetics and Depression: A systematic review. J. Neurosci. Biobehav. Rev. 2019, 102, 139–152. [Google Scholar] [CrossRef]
- Turecki, G.; Meaney, M.J. Effects of the social environment and stress on glucocorticoid receptor gene methylation: A systematic review. Biol. Psychiatry 2016, 79, 87–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakobsson, J.; Cordero, M.I.; Bisaz, R. KAP1-mediated epigenetic repression in the forebrain modulates behavioral vulnerability to stress. Neuron 2008, 60, 818–831. [Google Scholar] [CrossRef] [PubMed]
- Kyoung, S.M.; Ngoc, L.N.; Hong, L.C. Early life stress increases stress vulnerability through BDNF gene epigenetic changes in the rat hippocampus. Neuropharmacology 2016, 105, 388–397. [Google Scholar]
- The Network and Pathway Analysis Subgroup of the Psychiatric Genomics Consortium. Correction: Corrigendum: Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat. Neurosci. 2015, 18, 926. [Google Scholar] [CrossRef] [Green Version]
- Maes, M. Depression is an inflammatory disease, but cell-mediated immune activation is the key component of depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 664–675. [Google Scholar] [CrossRef]
- Maes, M. Evidence for an immune response in major depression: A review and hypothesis. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 1995, 19, 38. [Google Scholar] [CrossRef]
- Peng, Z.; Zhang, C.; Yan, L.; Zhang, Y.; Yang, Z.; Wang, J.; Song, C. EPA is more effective than DHA to improve depression-like behavior, glia cell dysfunction and hippcampal apoptosis signaling in a chronic stress-induced rat model of depression. Int. J. Mol. Sci. 2020, 21, 1769. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.I.; Song, D.K.; Shin, D.H. Associations of IL1RAP and IL1RL1 gene polymorphisms with obesity and inflammation mediators. Inflamm Res. 2020, 69, 191–202. [Google Scholar] [CrossRef]
- Kleiger, G.; Mayor, T. Perilous journey: A tour of the ubiquitin–proteasome system. Trends. Cell Biol. 2014, 24, 352–359. [Google Scholar] [CrossRef] [Green Version]
- Maure, J.F.; Moser, S.C.; Jaffray, E.G.; Alpi, A.F.; Hay, R.T. Loss of ubiquitin E2 Ube2w rescues hypersensitivity of Rnf4 mutant cells to DNA damage. Sci. Rep. 2016, 6, 26178. [Google Scholar] [CrossRef] [Green Version]
- Scaglione, K.M.; Zavodszky, E.; Todi, S.V.; Patury, S.; Xu, P.; Rodriguez-Lebron, E. Ube2w and ataxin-3 coordinately regulate the ubiquitin ligase CHIP. Mol. Cell 2011, 43, 599–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostafavi, S.; Battle, A.; Zhu, X.; Potash, J.B.; Weissman, M.M.; Shi, J. Type I interferon signaling genes in recurrent major depression: Increased expression detected by whole-blood RNA sequencing. Mol. Psychiatry 2014, 19, 1267–1274. [Google Scholar] [CrossRef] [PubMed]
- Leday, G.G.R.; Vértes, P.E.; Richardson, S. Replicable and coupled changes in innate and adaptive immune gene expression in two case-control studies of blood microarrays in major depressive disorder. Biol. Psychiatry 2018, 83, 70–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegel, I.; Salomon, D.; Erne, B.; Schaeren-Wiemers, N.; Peles, E. Caspr3 and caspr4, two novel members of the caspr family are expressed in the nervous system and interact with PDZ domains. Mol. Cell Neurosci. 2002, 20, 283–297. [Google Scholar] [CrossRef] [PubMed]
- Chua, S.E.; Cheung, C.; Cheung, V.; Tsang, J.T.; Chen, E.Y.; Wong, J.C. Cerebral grey white matter and csf in never-medicated, firstepisode schizophrenia. Schizophr. Res. 2007, 89, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Cordon, I.; Nicolas, M.J.; Arrieta, S.; Lopetegui, E.L.; Alegre, M. Coupling in the cortico-basal ganglia circuit is aberrant in the ketamine model of schizophrenia. Eur. Neuropsychopharmacol. 2015, 25, 1375–1387. [Google Scholar] [CrossRef]
- Hirata, H.; Takahashi, A.; Shimoda, Y.; Koide, T. Caspr3-deficient mice exhibit low motor learning during the early phase of the accelerated rotarod task. PLoS ONE 2016, 11, e0147887. [Google Scholar] [CrossRef] [Green Version]
- Kendler, K.S.; Gatz, M.; Gardner, C.O.; Pedersen, N.L. Clinical indices of familial depression in the swedish twin registry. Acta Psychiatr. Scand. 2007, 115, 214–220. [Google Scholar] [CrossRef]
- Kessler, R.C. The effects of stressful life events on depression. Annu. Rev. Psychol. 1997, 48, 191–214. [Google Scholar] [CrossRef]
- Wang, C.; Guo, J.; Guo, R. Effect of XingPiJieYu decoction on spatial learning and memory and cAMP-PKA-CREB-BDNF pathway in rat model of depression through chronic unpredictable stress. BMC Complementary Altern. Med. 2017, 17, 73. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, J.L.; Shapero, B.G.; Stange, J.P. Emotional maltreatment, peer victimization, and depressive versus anxiety symptoms during adolescence: Hopelessness as a mediator. J. Clin. Child. Adolesc. Psychol. 2013, 42, 332–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vugt, E.V.; Lanctt, N.; Paquette, G.; Collin-Vézina, D.; Lemieux, A. Girls in residential care: From child maltreatment to trauma-related symptoms in emerging adulthood. Child. Abus. Negl. 2014, 38, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Richards, E.M.; Niciu, M.J.; Ionescu, D.F.; Zoghbi, S.S.; Hong, J. cAMP signaling in brain is decreased in unmedicated depressed patients and increased by treatment with a selective serotonin reuptake inhibitor. Mol. Psychiatry 2017, 22, 754–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Xu, S.; Yuan, Z.; Shen, L. Weighted gene coexpression network analysis identifies specific modules and hub genes related to major depression. Neuropsychiatr. Dis Treat. 2020, 16, 703–713. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | HC (n = 10) | MDD (n = 10) |
|---|---|---|
| Age | 16.5(1.6) | 15.9(1.4) |
| Gender (male/female) | 4/6 | 4/6 |
| HDRS Mean (s.d) | 1.5(0.7) | 42.1(11.8) |
| PHQ-9 Mean (s.d) | 1.4(1.3) | 23.3(1.8) |
| Module Colors | Gene Number |
|---|---|
| black | 56 |
| blue | 215 |
| brown | 295 |
| green | 77 |
| magenta | 35 |
| pink | 41 |
| red | 58 |
| turquoise | 294 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, B.; Fan, Q.; Liu, J.; Yin, A.; Wang, P.; Zhang, W. Identification of Key Modules and Genes Associated with Major Depressive Disorder in Adolescents. Genes 2022, 13, 464. https://doi.org/10.3390/genes13030464
Zhao B, Fan Q, Liu J, Yin A, Wang P, Zhang W. Identification of Key Modules and Genes Associated with Major Depressive Disorder in Adolescents. Genes. 2022; 13(3):464. https://doi.org/10.3390/genes13030464
Chicago/Turabian StyleZhao, Bao, Qingyue Fan, Jintong Liu, Aihua Yin, Pingping Wang, and Wenxin Zhang. 2022. "Identification of Key Modules and Genes Associated with Major Depressive Disorder in Adolescents" Genes 13, no. 3: 464. https://doi.org/10.3390/genes13030464
APA StyleZhao, B., Fan, Q., Liu, J., Yin, A., Wang, P., & Zhang, W. (2022). Identification of Key Modules and Genes Associated with Major Depressive Disorder in Adolescents. Genes, 13(3), 464. https://doi.org/10.3390/genes13030464