
Spatiotemporal Regulation of Circular RNA Expression during Liver Development of Chinese Indigenous Ningxiang Pigs


Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Sample Collection
2.2. RNA Extraction
2.3. Library Preparation and Sequencing
2.4. Read Mapping and Transcriptome Assembly
2.5. Identification of circRNAs
2.6. Differential Expression Analysis and Functional Enrichment
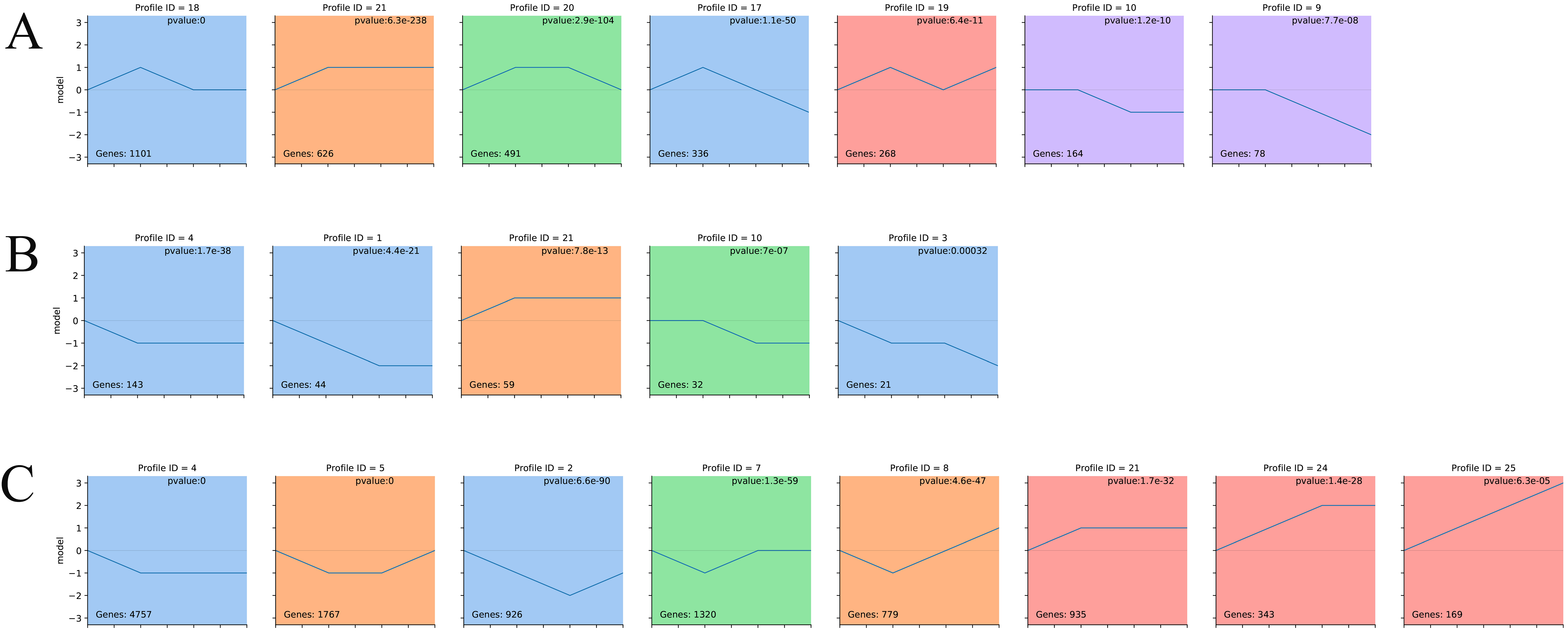
2.7. Time-Series Analysis
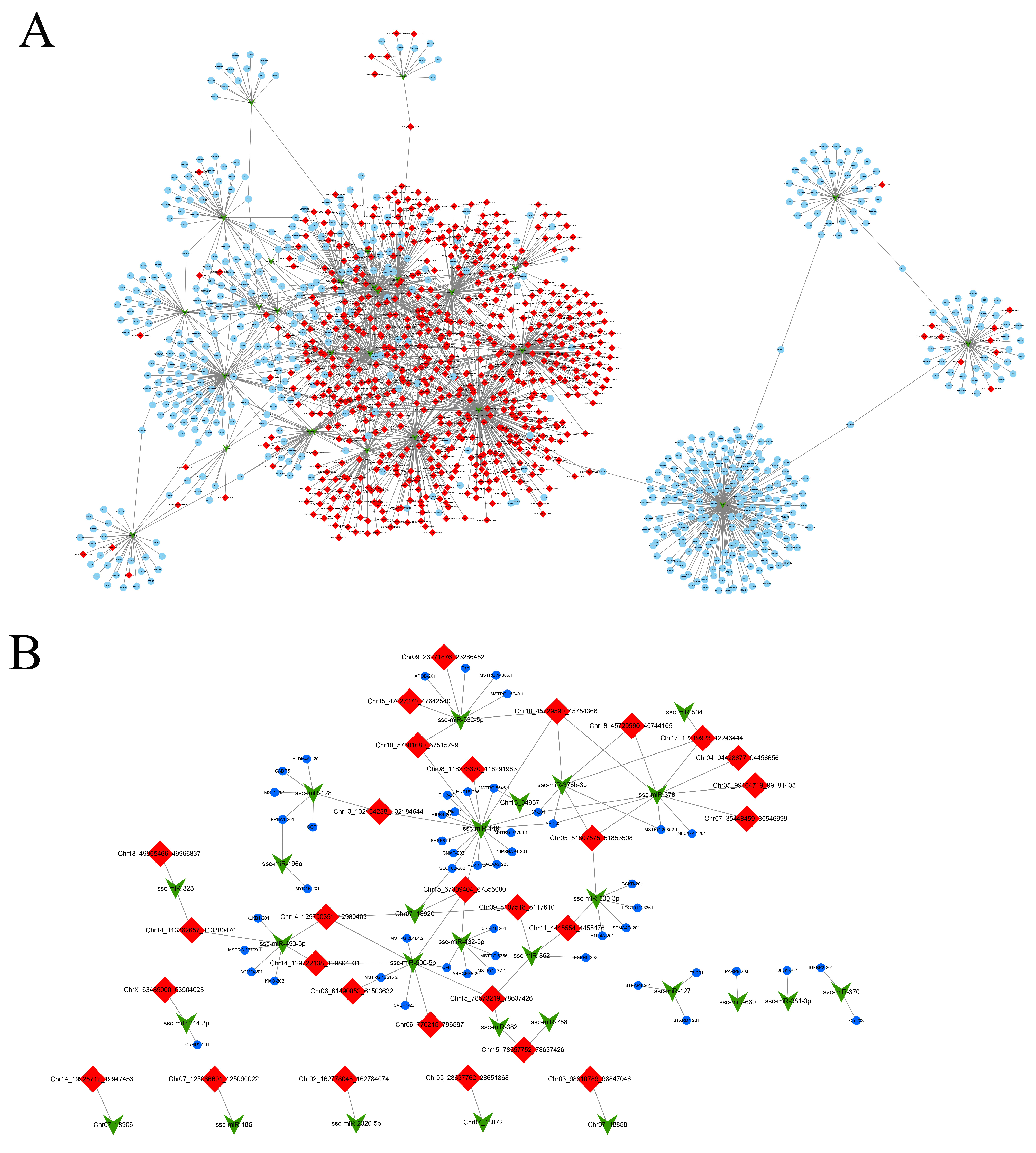
2.8. Analysis of ceRNAs Regulatory Networks
2.9. Validation of Expression by PCR
2.10. Statistical Analysis
3. Results
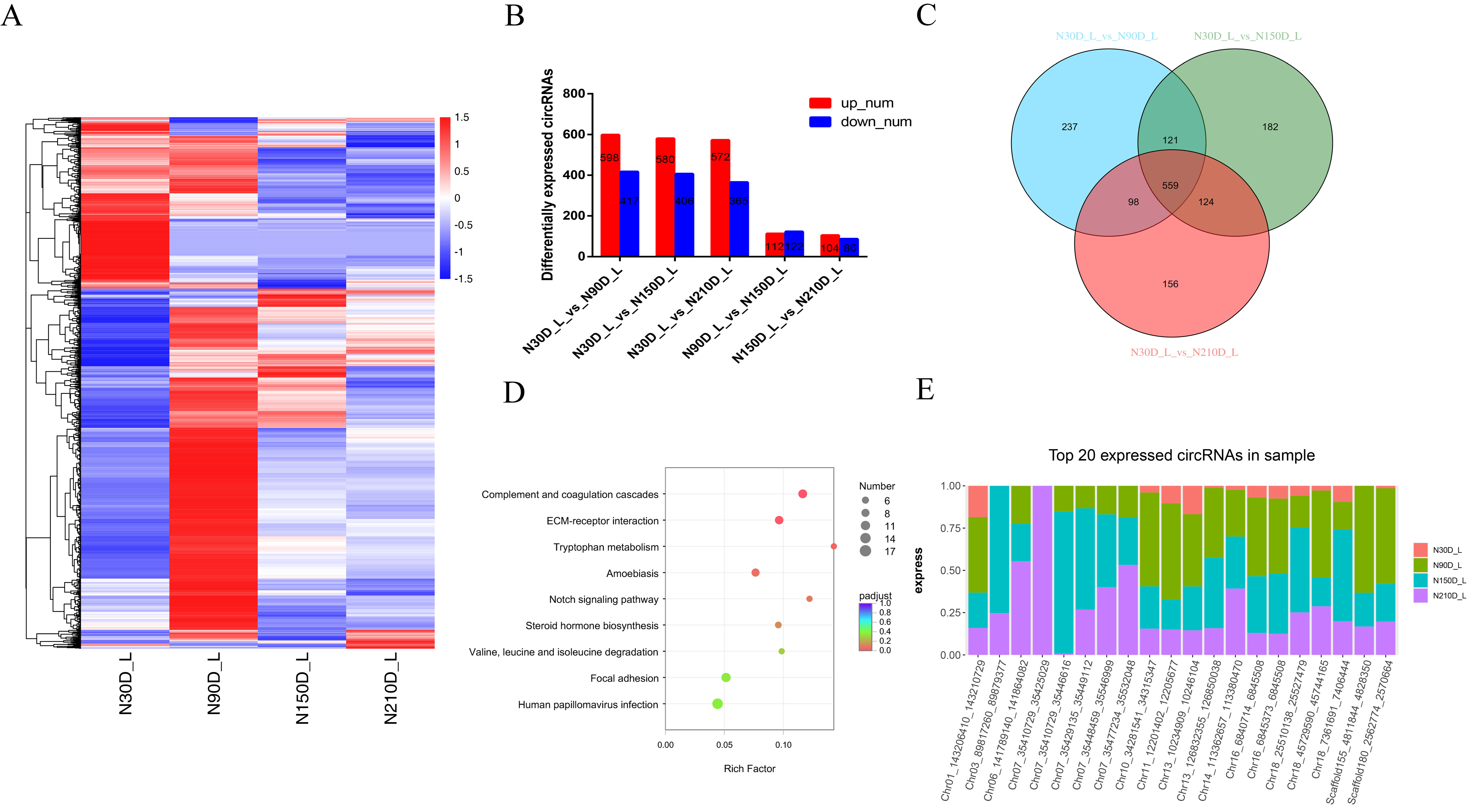
3.1. Identification and Characteristics of circRNAs in Ningxiang Pig Liver
3.2. Spatiotemporal Dynamic Expression Pattern of circRNAs in Liver of Ningxiang Pigs
3.3. Constructing the circRNA–miRNA–mRNA Coexpression Networks through Time-Series Analysis
3.4. RT-qPCR Quantification of circRNAs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, F.; Gao, H.; Zhang, Y.; Liao, Y.; Zeng, Q.; He, X.; Xu, K.; He, J. Optimizing conditions of electronic nose for rapid detection of flavor substances in Ningxiang Pork. J. Food Process Eng. 2021, 44, e13758. [Google Scholar] [CrossRef]
- Ma, H.; Jiang, J.; He, J.; Huifang, L.; Han, L.; Gong, Y.; Li, B.; Zonggang, Y.; Shengguo, T.; Zhang, Y.; et al. Long-read assembly of the Chinese indigenous Ningxiang pig genome and identification of genetic variations in fat metabolism among different breeds. Mol. Ecol. Resour. 2021. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Ren, P.; Kong, X.; Wu, Y.; Wu, G.; Li, P.; Hao, F.; Tang, H.; Blachier, F.; Yin, Y. Comparison of serum metabolite compositions between obese and lean growing pigs using an NMR-based metabonomic approach. J. Nutr. Biochem. 2012, 23, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Tao, C.; Xu, J.; Ruan, J.; Xia, J.; Zhu, W.; Xin, L.; Ye, H.; Xie, N.; Xia, B.; et al. CD8 T Cells Involved in Metabolic Inflammation in Visceral Adipose Tissue and Liver of Transgenic Pigs. Front. Immunol. 2021, 12, 690069. [Google Scholar] [CrossRef]
- Ahn, J.; Lee, H.; Chung, C.; Ha, T. High fat diet induced downregulation of microRNA-467b increased lipoprotein lipase in hepatic steatosis. Biochem. Biophys. Res. Commun. 2011, 414, 664–669. [Google Scholar] [CrossRef]
- Smolka, C.; Schlösser, D.; Hohnloser, C.; Bemtgen, X.; Jänich, C.; Schneider, L.; Martin, J.; Pfeifer, D.; Moser, M.; Hasselblatt, P.; et al. MiR-100 overexpression attenuates high fat diet induced weight gain, liver steatosis, hypertriglyceridemia and development of metabolic syndrome in mice. Mol. Med. 2021, 27, 101. [Google Scholar] [CrossRef]
- Wang, J.; Ren, Q.; Hua, L.; Chen, J.; Zhang, J.; Bai, H.; Li, H.; Xu, B.; Shi, Z.; Cao, H.; et al. Comprehensive Analysis of Differentially Expressed mRNA, lncRNA and circRNA and Their ceRNA Networks in the Longissimus Dorsi Muscle of Two Different Pig Breeds. Int. J. Mol. Sci. 2019, 20, 1107. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.; Zhang, Y.; Han, B.; Bai, Y.; Zhou, R.; Gan, G.; Chao, J.; Hu, G.; Yao, H. Circular RNA HIPK2 regulates astrocyte activation via cooperation of autophagy and ER stress by targeting MIR124-2HG. Autophagy 2017, 13, 1722–1741. [Google Scholar] [CrossRef] [Green Version]
- Zang, J.; Lu, D.; Xu, A. The interaction of circRNAs and RNA binding proteins: An important part of circRNA maintenance and function. J. Neurosci. Res. 2020, 98, 87–97. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Zhao, J.; Zhao, Y.; Li, W.; Zhao, L.; Ren, Y.; Ou, R.; Xu, Y. Hsa_circ_0048179 attenuates free fatty acid-induced steatosis via hsa_circ_0048179/miR-188-3p/GPX4 signaling. Aging 2020, 12, 23996–24008. [Google Scholar] [CrossRef]
- Yu, G.; Yang, Z.; Peng, T.; Lv, Y. Circular RNAs: Rising stars in lipid metabolism and lipid disorders. J. Cell. Physiol. 2021, 236, 4797–4806. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Yang, J.; He, J.; Gong, Y.; Xiao, Y.; Zeng, Q.; Xu, K.; Duan, Y.; He, J.; Ma, H. Spatiotemporal Regulation and Functional Analysis of Circular RNAs in Skeletal Muscle and Subcutaneous Fat during Pig Growth. Biology 2021, 10, 841. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Li, P.; Zhou, W.; Fan, L.; Wang, B.; Liu, H.; Gao, C.; Du, T.; Pu, G.; Wu, C.; et al. Effects of various levels of dietary fiber on carcass traits, meat quality and myosin heavy chain I, IIa, IIx and IIb expression in muscles in Erhualian and Large White pigs. Meat Sci. 2020, 169, 108160. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Bar-Joseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinform. 2006, 7, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Zhou, L.; He, Z.; Chen, Y.; Jiang, X.; Xu, J.; Jiang, J. Circular RNA hsa_circ_0006117 Facilitates Pancreatic Cancer Progression by Regulating the miR-96-5p/KRAS/MAPK Signaling Pathway. J. Oncol. 2021, 2021, 9213205. [Google Scholar] [CrossRef]
- Gong, Y.; Zhang, Y.; Li, B.; Xiao, Y.; Zeng, Q.; Xu, K.; Duan, Y.; He, J.; Ma, H. Insight into Liver lncRNA and mRNA Profiling at Four Developmental Stages in Ningxiang Pig. Biology 2021, 10, 310. [Google Scholar] [CrossRef]
- Huang, Y.; Ge, W.; Ding, Y.; Zhang, L.; Zhou, J.; Kong, Y.; Cui, B.; Gao, B.; Qian, X.; Wang, W. The circular RNA circSLC7A11 functions as a mir-330-3p sponge to accelerate hepatocellular carcinoma progression by regulating cyclin-dependent kinase 1 expression. Cancer Cell Int. 2021, 21, 636. [Google Scholar] [CrossRef]
- Kim, E.; Kim, Y.K.; Lee, S.-J.V. Emerging functions of circular RNA in aging. Trends Genet. 2021, 37, 819–829. [Google Scholar] [CrossRef]
- Krishnamoorthy, A.; Kadener, S. Using Drosophila to uncover molecular and physiological functions of circRNAs. Methods 2021, 196, 74–84. [Google Scholar] [CrossRef]
- Liang, G.; Yang, Y.; Niu, G.; Tang, Z.; Li, K. Genome-wide profiling of Sus scrofa circular RNAs across nine organs and three developmental stages. DNA Res. 2017, 24, 523–535. [Google Scholar] [CrossRef] [Green Version]
- Venø, M.T.; Hansen, T.B.; Venø, S.T.; Clausen, B.H.; Grebing, M.; Finsen, B.; Holm, I.E.; Kjems, J. Spatio-temporal regulation of circular RNA expression during porcine embryonic brain development. Genome Biol. 2015, 16, 245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, G.; Zhang, H.; Li, X.; Hu, J.; Yang, G.; Sun, S. Genome-Wide Differential Expression Profiling of Ovarian circRNAs Associated With Litter Size in Pigs. Front. Genet. 2019, 10, 1010. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S. Role and mechanism of action of angiopoietin-like protein ANGPTL4 in plasma lipid metabolism. J. Lipid Res. 2021, 62, 100150. [Google Scholar] [CrossRef] [PubMed]
- Kocks, C.; Boltengagen, A.; Piwecka, M.; Rybak-Wolf, A.; Rajewsky, N. Single-Molecule Fluorescence In Situ Hybridization (FISH) of Circular RNA CDR1as. Methods Mol. Biol. 2018, 1724, 77–96. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Vélez, R.; Izquierdo, M.; Correa-Bautista, J.; Correa-Rodríguez, M.; Schmidt-RioValle, J.; González-Jiménez, E.; González-Jiménez, K. Liver Fat Content and Body Fat Distribution in Youths with Excess Adiposity. J. Clin. Med. 2018, 7, 528. [Google Scholar] [CrossRef] [Green Version]
- Duran, J.; Navarro-Sabate, A.; Pujol, A.; Perales, J.C.; Manzano, A.; Obach, M.; Gómez, M.; Bartrons, R. Overexpression of ubiquitous 6-phosphofructo-2-kinase in the liver of transgenic mice results in weight gain. Biochem. Biophys. Res. Commun. 2008, 365, 291–297. [Google Scholar] [CrossRef]
- Demirkan, A.; van Duijn, C.M.; Ugocsai, P.; Isaacs, A.; Pramstaller, P.P.; Liebisch, G.; Wilson, J.F.; Johansson, Å.; Rudan, I.; Aulchenko, Y.S.; et al. Genome-wide association study identifies novel loci associated with circulating phospho- and sphingolipid concentrations. PLoS Genet. 2012, 8, e1002490. [Google Scholar] [CrossRef] [Green Version]
- Nilsen, M.S.; Jersin, R.Å.; Ulvik, A.; Madsen, A.; McCann, A.; Svensson, P.-A.; Svensson, M.K.; Nedrebø, B.G.; Gudbrandsen, O.A.; Tell, G.S.; et al. 3-Hydroxyisobutyrate, A Strong Marker of Insulin Resistance in Type 2 Diabetes and Obesity That Modulates White and Brown Adipocyte Metabolism. Diabetes 2020, 69, 1903–1916. [Google Scholar] [CrossRef]
- Dong, X.; Zhu, Y.; Wang, S.; Luo, Y.; Lu, S.; Nan, F.; Sun, G.; Sun, X. Bavachinin inhibits cholesterol synthesis enzyme FDFT1 expression via AKT/mTOR/SREBP-2 pathway. Int. Immunopharmacol. 2020, 88, 106865. [Google Scholar] [CrossRef]
- Cadenas, C.; Vosbeck, S.; Hein, E.-M.; Hellwig, B.; Langer, A.; Hayen, H.; Franckenstein, D.; Büttner, B.; Hammad, S.; Marchan, R.; et al. Glycerophospholipid profile in oncogene-induced senescence. Biochim. Biophys. Acta 2012, 1821, 1256–1268. [Google Scholar] [CrossRef]
- Yudkin, J.S.; Stehouwer, C.D.; Emeis, J.J.; Coppack, S.W. C-reactive protein in healthy subjects: Associations with obesity, insulin resistance, and endothelial dysfunction: A potential role for cytokines originating from adipose tissue? Arterioscler. Thromb. Vasc. Biol. 1999, 19, 972–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pou, K.M.; Massaro, J.M.; Hoffmann, U.; Vasan, R.S.; Maurovich-Horvat, P.; Larson, M.G.; Keaney, J.F., Jr.; Meigs, J.B.; Lipinska, I.; Kathiresan, S.; et al. Visceral and subcutaneous adipose tissue volumes are cross-sectionally related to markers of inflammation and oxidative stress: The Framingham Heart Study. Circulation 2007, 116, 1234–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiScipio, R.G.; Chakravarti, D.N.; Muller-Eberhard, H.J.; Fey, G.H. The structure of human complement component C7 and the C5b-7 complex. J. Biol. Chem. 1988, 263, 549–560. [Google Scholar] [CrossRef]
- Van der Meer, B.W.; Fugate, R.D.; Sims, P.J. Complement proteins C5b-9 induce transbilayer migration of membrane phospholipids. Biophys. J. 1989, 56, 935–946. [Google Scholar] [CrossRef] [Green Version]
- Heo, J.; Lee, S.; Jo, S.; Ko, J.; Kwon, H.; Hong, E. Hepatic LKB1 Reduces the Progression of Non-Alcoholic Fatty Liver Disease via Genomic Androgen Receptor Signaling. Int. J. Mol. Sci. 2021, 22, 7904. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Inukai, K.; Ikegami, Y.; Awata, T.; Katayama, S. LKB1, an upstream AMPK kinase, regulates glucose and lipid metabolism in cultured liver and muscle cells. Biochem. Biophys. Res. Commun. 2006, 351, 595–601. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Terms | NX30d | NX90d | NX150d | NX210d |
|---|---|---|---|---|
| Raw reads number | 56500489 | 55531984 | 45030983 | 51897636 |
| Clean reads number | 55574608 | 54755520 | 44381475 | 51069717 |
| Clean reads rate | 98.36% | 98.60% | 98.56% | 98.40% |
| Clean Q30 bases rate | 95.14% | 95.55% | 95.28% | 95.52% |
| Mapped reads | 102827570 | 103499271 | 83368071 | 95480783 |
| Mapping rate | 92.51% | 94.51% | 93.92% | 93.48 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, W.; Ma, H.; Li, B.; Yang, F.; Xiao, Y.; Gong, Y.; Li, Z.; Li, T.; Zeng, Q.; Xu, K.; et al. Spatiotemporal Regulation of Circular RNA Expression during Liver Development of Chinese Indigenous Ningxiang Pigs. Genes 2022, 13, 746. https://doi.org/10.3390/genes13050746
Chen W, Ma H, Li B, Yang F, Xiao Y, Gong Y, Li Z, Li T, Zeng Q, Xu K, et al. Spatiotemporal Regulation of Circular RNA Expression during Liver Development of Chinese Indigenous Ningxiang Pigs. Genes. 2022; 13(5):746. https://doi.org/10.3390/genes13050746
Chicago/Turabian StyleChen, Wenwu, Haiming Ma, Biao Li, Fang Yang, Yu Xiao, Yan Gong, Zhi Li, Ting Li, Qinghua Zeng, Kang Xu, and et al. 2022. "Spatiotemporal Regulation of Circular RNA Expression during Liver Development of Chinese Indigenous Ningxiang Pigs" Genes 13, no. 5: 746. https://doi.org/10.3390/genes13050746
APA StyleChen, W., Ma, H., Li, B., Yang, F., Xiao, Y., Gong, Y., Li, Z., Li, T., Zeng, Q., Xu, K., & Duan, Y. (2022). Spatiotemporal Regulation of Circular RNA Expression during Liver Development of Chinese Indigenous Ningxiang Pigs. Genes, 13(5), 746. https://doi.org/10.3390/genes13050746