
The Complete Mitochondrial Genome of Stichopus naso (Aspidochirotida: Stichopodidae: Stichopus) and Its Phylogenetic Position

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Mitogenome Sequencing
2.3. Sequence Annotation and Analysis
2.4. Phylogenetic Analysis
3. Results and Discussion
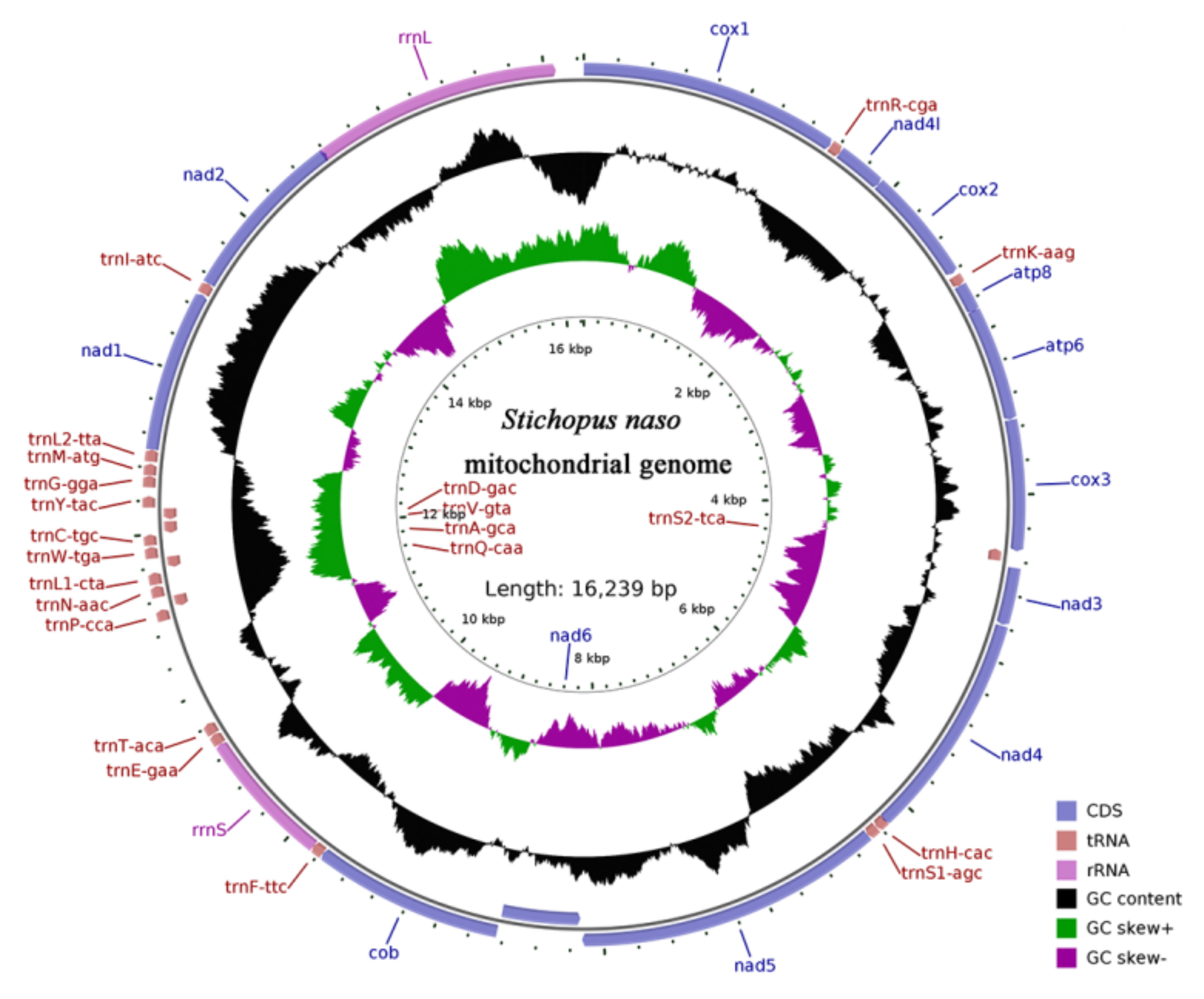
3.1. Genome Organization and Base Composition
3.2. Protein-Coding Genes and Codon Usage
3.3. Transfer RNA and Ribosomal RNA Genes
3.4. Gene Rearrangement
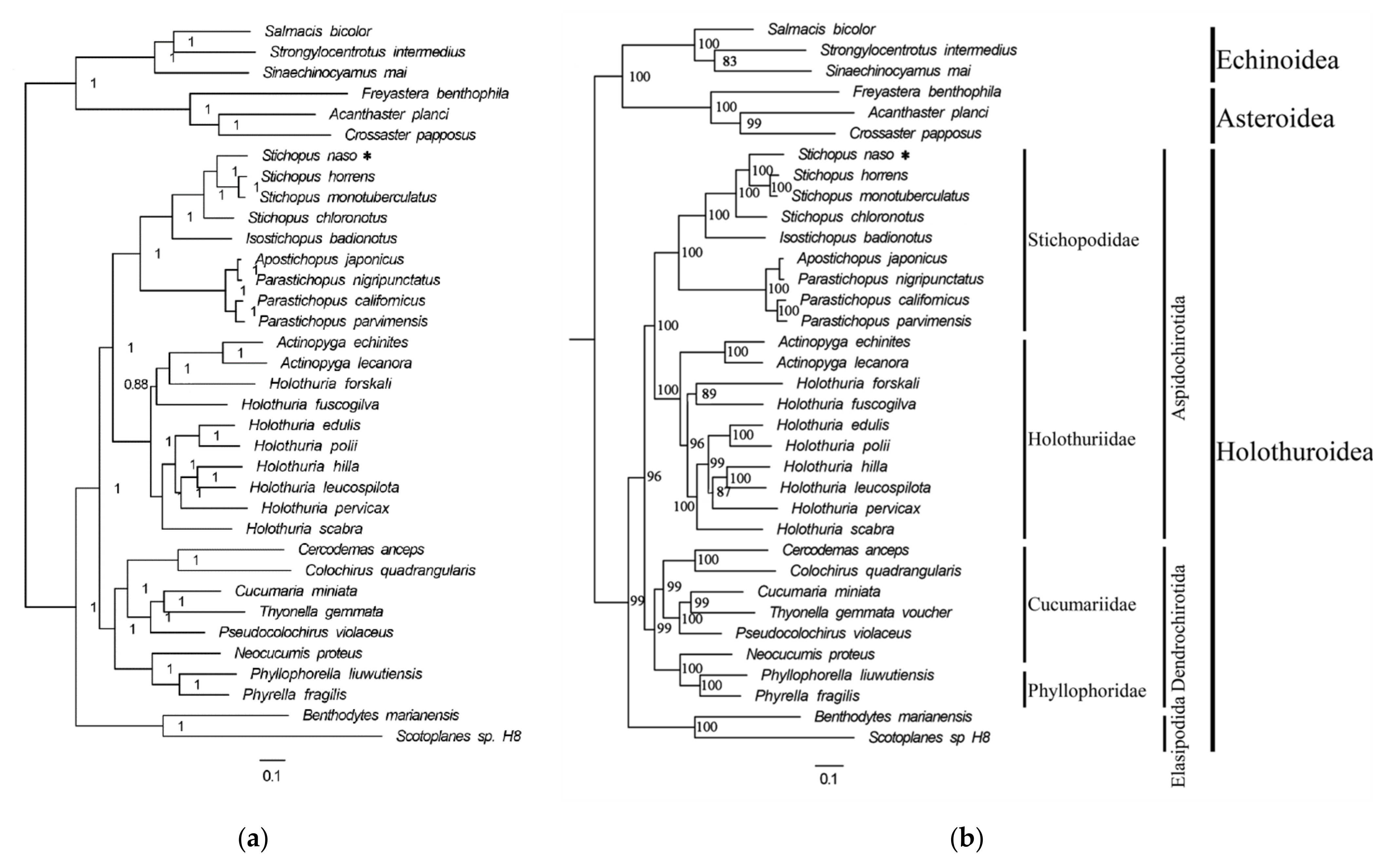
3.5. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Caccone, A.; Gentile, G.; Burns, C.E.; Sezzi, E.; Bergman, W.; Ruelle, M.; Saltonstall, K.; Powell, J.R. Extreme difference in rate of mitochondrial and nuclear DNA evolution in a large ectotherm, Galapagos tortoises. Mol. Phylogenet. Evol. 2004, 31, 794–798. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Gong, L.; Lu, X.; Jiang, L.; Liu, B.; Liu, L.; Lue, Z.; Li, P.; Zhang, X. Gene rearrangements in the mitochondrial genome of Chiromantes eulimene (Brachyura: Sesarmidae) and phylogenetic implications for Brachyura. Int. J. Biol. Macromol. 2020, 162, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Rubinoff, D.; Holland, B.S. Between two extremes: Mitochondrial DNA is neither the panacea nor the nemesis of phylogenetic and taxonomic inference. Syst. Biol. 2005, 54, 952–961. [Google Scholar] [CrossRef]
- Wen, J.; Hu, C.; Zhang, L.; Fan, S. Genetic identification of global commercial sea cucumber species on the basis of mitochondrial DNA sequences. Food Control. 2011, 22, 72–77. [Google Scholar] [CrossRef]
- Kerr, A.M.; Kim, J.H. Phylogeny of Holothuroidea (Echinodermata) inferred from morphology. J. Linn. Soc. Lond. Zool. 2001, 133, 63–81. [Google Scholar] [CrossRef]
- Gallo, N.D.; Cameron, J.; Hardy, K.; Fryer, P.; Bartlett, D.H.; Levin, L.A. Submersible- and lander-observed community patterns in the Mariana and New Britain trenches: Influence of productivity and depth on epibenthic and scavenging communities. Deep. Sea Res. Part I 2015, 99, 119–133. [Google Scholar] [CrossRef] [Green Version]
- Prata, J.; Manso, C.L.C.; Christoffersen, M.L. Aspidochirotida (Echinodermata: Holothuroidea) from the northeast coast of Brazil. Zootaxa 2014, 3889, 127–150. [Google Scholar] [CrossRef]
- Rowe, F.W.E.; Richmond, M.D. A preliminary account of the shallow-water echinoderms of Rodrigues, Mauritius, western Indian Ocean. J. Nat. Hist. 2004, 38, 3273–3314. [Google Scholar] [CrossRef]
- Samyn, Y.; Tallon, I. Zoogeography of the shallow-water holothuroids of the western Indian Ocean. J. Biogeogr. 2005, 32, 1523–1538. [Google Scholar] [CrossRef]
- Wolfe, K.; Vidal-Ramirez, F.; Dove, S.; Deaker, D.; Byrne, M. Altered sediment biota and lagoon habitat carbonate dynamics due to sea cucumber bioturbation in a high-pCO(2) environment. GCB Bioenergy 2018, 24, 465–480. [Google Scholar] [CrossRef]
- Tuwo, A.; Tresnati, J. Sea Cucumber Farming in Southeast Asia (Malaysia, Philippines, Indonesia, Vietnam); Wiley: Hoboken, NJ, USA, 2015; pp. 331–352. [Google Scholar]
- Byrne, M.; Rowe, F.; Uthicke, S. Molecular taxonomy, phylogeny and evolution in the family Stichopodidae (Aspidochirotida: Holothuroidea) based on COI and 16S mitochondrial DNA. Mol. Phylogenet. Evol. 2010, 56, 1068–1081. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Ma, C.; Ma, L. Molecular identification of genus Scylla (Decapoda: Portunidae) based on DNA barcoding and polymerase chain reaction. Biochem. Syst. Ecol. 2012, 41, 41–47. [Google Scholar] [CrossRef]
- Nishihama, S.; Yamana, Y.; Yoshimura, T. First record of the tropical holothurian Stichopus naso Semper, 1867 (Echinodermata: Holothuroidea: Synallactida) from the temperate coast of Kyushu mainland, Japan, in relation to ocean warming. Plankton Benthos. Res. 2020, 15, 66–71. [Google Scholar] [CrossRef] [Green Version]
- Massin, C. Redescription of Stichopus naso Semper, 1868 (Echinodermata, Holothuroidea, Stichopodidae). Bull. L’institut R. Sci. Nat. Belg. 2007, 77, 123–130. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernt, M.; Donath, A.; Juehling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Puetz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef]
- Edgar, R.C. Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kueck, P.; Meusemann, K. FASconCAT: Convenient handling of data matrices. Mol. Phylogenet. Evol. 2010, 56, 1115–1118. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Xie, Z. DAMBE: Software package for data analysis in molecular biology and evolution. J. Hered. 2001, 92, 371–373. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Fan, S.; Hu, C.; Wen, J.; Zhang, L. Characterization of mitochondrial genome of sea cucumber Stichopus horrens: A novel gene arrangement in Holothuroidea. Sci. China-Life Sci. 2011, 54, 434–441. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Sun, Y.; Zhao, H.; Hu, J.; Chen, B.; Li, H.; Huang, W. Complete mitochondrial genome of a tropical sea cucumber, Stichopus chloronotus. Mitochondrial DNA B 2021, 6, 2788–2790. [Google Scholar] [CrossRef]
- Utzeri, V.J.; Ribani, A.; Bovo, S.; Taurisano, V.; Calassanzio, M.; Baldo, D.; Fontanesi, L. Microscopic ossicle analyses and the complete mitochondrial genome sequence of Holothuria (Roweothuria) polii (Echinodermata; Holothuroidea) provide new information to support the phylogenetic positioning of this sea cucumber species. Mar. Genom. 2020, 51, 100735. [Google Scholar] [CrossRef]
- Yang, Q.; Lin, Q.; Wu, J.; Ngoc Tuan, T.; Huang, R.; Sun, Z.; Zhu, Z.; Lu, Z.; Li, S.; Zhou, C. Complete mitochondrial genome of Holothuria leucospilata (Holothuroidea, Holothuriidae) and phylogenetic analysis. Mitochondrial DNA B 2019, 4, 2751–2752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Tang, D.; Guo, H.; Wang, J.; Xu, X.; Wang, Z. Comparative mitochondrial genomic analysis of Macrophthalmus pacificus and insights into the phylogeny of the Ocypodoidea & Grapsoidea. Genomics 2020, 112, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Francino, M.P.; Ochman, H. Strand asymmetries in DNA evolution. Trends Genet. 1997, 13, 240–245. [Google Scholar] [CrossRef]
- Reyes, A.; Gissi, C.; Pesole, G.; Saccone, C. Asymmetrical directional mutation pressure in the mitochondrial genome of mammals. Mol. Biol. Evol. 1998, 15, 957–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, X. DNA Replication and Strand Asymmetry in Prokaryotic and Mitochondrial Genomes. Curr. Genom. 2012, 13, 16–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, W.; Liu, J.; Zhang, H. Complete mitochondrial genome of Benthodytes marianensis (Holothuroidea: Elasipodida: Psychropotidae): Insight into deep sea adaptation in the sea cucumber. PLoS ONE 2018, 13, e0208051. [Google Scholar] [CrossRef]
- Fan, S.; Hu, C.; Zhang, L.; Sun, H.; Wen, J.; Luo, P. Complete mitochondrial genome of the sea cucumber Stichopus sp and its application in the identification of this species. Aquacult. Res. 2012, 43, 1306–1316. [Google Scholar] [CrossRef]
- Yang, F.; Zhou, C.; Tran, N.T.; Sun, Z.; Wu, J.; Ge, H.; Lu, Z.; Zhong, C.; Zhu, Z.; Yang, Q.; et al. Comparison of the complete mitochondrial genome of Phyllophorus liuwutiensis (Echinodermata: Holothuroidea: Phyllophoridae) to that of other sea cucumbers. FEBS Open Bio. 2020, 10, 1587–1600. [Google Scholar] [CrossRef]
- Shen, X.; Tian, M.; Liu, Z.; Cheng, H.; Tan, J.; Meng, X.; Ren, J. Complete mitochondrial genome of the sea cucumber Apostichopus japonicus (Echinodermata: Holothuroidea): The first representative from the subclass Aspidochirotacea with the echinoderm ground pattern. Gene 2009, 439, 79–86. [Google Scholar] [CrossRef]
- Sun, S.; Sha, Z.; Xiao, N. The first two complete mitogenomes of the order Apodida from deep-sea chemoautotrophic environments: New insights into the gene rearrangement, origin and evolution of the deep-sea sea cucumbers. Comp. Biochem. Physiol. Part D Genom. Proteom. 2021, 39, 100839. [Google Scholar] [CrossRef]
- Lin, J.-P.; Tsai, M.-H.; Kroh, A.; Trautman, A.; Machado, D.J.; Chang, L.-Y.; Reid, R.; Lin, K.-T.; Bronstein, O.; Lee, S.-J.; et al. The first complete mitochondrial genome of the sand dollar Sinaechinocyamus mai (Echinoidea: Clypeasteroida). Genomics 2020, 112, 1686–1693. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Bae, Y.J.; Shin, S. Mitochondrial gene rearrangement and phylogenetic relationships in the Amphilepidida and Ophiacanthida (Echinodermata, Ophiuroidea). Mar. Biol. Res. 2019, 15, 26–35. [Google Scholar] [CrossRef]
- Sharp, P.M.; Li, W.H. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 1986, 24, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Li, X.; Sha, Z.; Yan, B.; Xu, Q. Complete mitochondrial genome of the Japanese snapping shrimp Alpheus japonicus (Crustacea: Decapoda: Caridea): Gene rearrangement and phylogeny within Caridea. Sci. China-Life Sci. 2012, 55, 591–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Shi, X.; Guo, H.; Tang, D.; Bai, Y.; Wang, Z. Characterization of the complete mitochondrial genome of Uca lacteus and comparison with other Brachyuran crabs. Genomics 2020, 112, 10–19. [Google Scholar] [CrossRef]
- Luis Teixeira, D.C.; Powell, C.; van Noort, S.; Costa, C.; Sinno, M.; Caleca, V.; Rhode, C.; Kennedy, R.J.; van Staden, M.; van Asch, B. The complete mitochondrial genome of Bactrocera biguttula (Bezzi) (Diptera: Tephritidae) and phylogenetic relationships with other Dacini. Int. J. Biol. Macromol. 2019, 126, 130–140. [Google Scholar] [CrossRef]
- Su, T.; Liang, A. Comparative analysis of seven mitochondrial genomes of Phymatostetha (Hemiptera: Cercopidae) and phylogenetic implications. Int. J. Biol. Macromol. 2019, 125, 1112–1117. [Google Scholar] [CrossRef]
- Kundu, S.; Kumar, V.; Tyagi, K.; Chakraborty, R.; Singha, D.; Rahaman, I.; Pakrashi, A.; Chandra, K. Complete mitochondrial genome of Black Soft-shell Turtle (Nilssonia nigricans) and comparative analysis with other Trionychidae. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Krebes, L.; Bastrop, R. The mitogenome of Gammarus duebeni (Crustacea Amphipoda): A new gene order and non-neutral sequence evolution of tandem repeats in the control region. Comp. Biochem. Physiol. Part D Genom. Proteom. 2012, 7, 201–211. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, P.-D.; Zhang, D.-Z.; Zhang, H.-B.; Tang, B.-P.; Liu, Q.-N.; Dai, L.-S. Mitochondrial genome of the yellow catfish Pelteobagrus fulvidraco and insights into Bagridae phylogenetics. Genomics 2019, 111, 1258–1265. [Google Scholar] [CrossRef]
- Juehling, T.; Duchardt-Ferner, E.; Bonin, S.; Woehnert, J.; Puetz, J.; Florentz, C.; Betat, H.; Sauter, C.; Moerl, M. Small but large enough: Structural properties of armless mitochondrial tRNAs from the nematode Romanomermis culicivorax. Nucleic Acids Res. 2018, 46, 9170–9180. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.-I.; Suematsu, T.; Ohtsuki, T. Losing the stem-loop structure from metazoan mitochondrial tRNAs and co-evolution of interacting factors. Front. Genet. 2014, 5, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montelli, S.; Peruffo, A.; Patarnello, T.; Cozzi, B.; Negrisolo, E. Back to Water: Signature of Adaptive Evolution in Cetacean Mitochondrial tRNAs. PLoS ONE 2016, 11, e0158129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Li, Q.; Fu, R.; Wang, J.; Deng, G.; Chen, X.; Lu, D. Comparative mitochondrial genome analysis reveals intron dynamics and gene rearrangements in two Trametes species. Sci. Rep. 2021, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Su, T.; He, B.; Li, K.; Liang, A. Comparative analysis of the mitochondrial genomes of oriental spittlebug trible Cosmoscartini: Insights into the relationships among closely related taxa. BMC Genom. 2018, 19, 961. [Google Scholar] [CrossRef]
- Arndt, A.; Smith, M.J. Mitochondrial gene rearrangement in the sea cucumber genus Cucumaria. Mol. Biol. Evol. 1998, 15, 1009–1016. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Zhu, K.; Liu, Y.; Zhang, H.; Gong, L.; Jiang, L.; Liu, L.; Lu, Z.; Liu, B. Novel gene rearrangement in the mitochondrial genome of Muraenesox cinereus and the phylogenetic relationship of Anguilliformes. Sci. Rep. 2021, 11, 1–14. [Google Scholar] [CrossRef]
- Uthicke, S.; Byrne, M.; Conand, C. Genetic barcoding of commercial Beche-de-mer species (Echinodermata: Holothuroidea). Mol. Ecol. Resour. 2010, 10, 634–646. [Google Scholar] [CrossRef]
- Miller, A.K.; Kerr, A.M.; Paulay, G.; Reich, M.; Wilson, N.G.; Carvajal, J.I.; Rouse, G.W. Molecular phylogeny of extant Holothuroidea (Echinodermata). Mol. Phylogenet. Evol. 2017, 111, 110–131. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Strand | Location | Size | Anticodon | Codon | Continuity | |
|---|---|---|---|---|---|---|---|
| Start | Stop | ||||||
| cox1 | + | 1-1554 | 1554 | ATG | TAA | 167 | |
| trnR | + | 1560-1627 | 68 | TCG | 5 | ||
| nad4l | + | 1628-1924 | 297 | ATG | TAA | 0 | |
| cox2 | + | 1925-2614 | 690 | ATG | TAA | 0 | |
| trnK | + | 2635-2698 | 64 | CTT | 20 | ||
| atp8 | + | 2699-2875 | 177 | ATG | TAA | 0 | |
| atp6 | + | 2869-3552 | 684 | ATG | TAA | −7 | |
| cox3 | + | 3555-4337 | 783 | ATG | TAA | 2 | |
| trnS2 | − | 4336-4406 | 71 | TGA | −2 | ||
| nad3 | + | 4438-4782 | 345 | GTG | TAG | 31 | |
| nad4 | + | 4790-6157 | 1368 | ATG | TAG | 7 | |
| trnH | + | 6148-6215 | 68 | GTG | −10 | ||
| trnS1 | + | 6218-6285 | 68 | GCT | 2 | ||
| nad5 | + | 6286-8130 | 1845 | ATG | TAA | 0 | |
| nad6 | − | 8143-8631 | 489 | ATG | TAA | 12 | |
| cob | + | 8640-9782 | 1143 | ATG | TAA | 8 | |
| trnF | + | 9783-9853 | 71 | GAA | 0 | ||
| rrnS | + | 9852-10678 | 827 | −2 | |||
| trnE | + | 10678-10746 | 69 | TTC | −1 | ||
| trnT | + | 10749-10818 | 70 | TGT | 2 | ||
| trnP | + | 11483-11550 | 68 | TGG | 664 | ||
| trnQ | − | 11547-11616 | 70 | TTG | −4 | ||
| trnN | + | 11626-11694 | 69 | GTT | 9 | ||
| trnL1 | + | 11703-11774 | 72 | TAG | 8 | ||
| trnA | − | 11791-11858 | 68 | TGC | 16 | ||
| trnW | + | 11859-11928 | 70 | TCA | 0 | ||
| trnC | + | 11937-12002 | 66 | GCA | 8 | ||
| trnV | − | 12004-12075 | 72 | TAC | 1 | ||
| trnD | − | 12089-12156 | 68 | GTC | 13 | ||
| trnY | + | 12163-12230 | 68 | GTA | 6 | ||
| trnG | + | 12283-12351 | 69 | TCC | 52 | ||
| trnM | + | 12354-12422 | 69 | CAT | 2 | ||
| trnL2 | + | 12436-12507 | 72 | TAA | 13 | ||
| nad1 | + | 12508-13479 | 972 | ATG | TAG | 0 | |
| trnI | + | 13487-13554 | 68 | GAT | 7 | ||
| nad2 | + | 13555-14601 | 1047 | GTG | TAA | 0 | |
| rrnL | + | 14577-16072 | 1496 | −25 | |||
| Region | A% | T% | G% | C% | A + T (%) | G + C (%) | AT-Skew | GC-Skew |
|---|---|---|---|---|---|---|---|---|
| mitogenome | 30.07 | 28.69 | 17.36 | 23.88 | 58.76 | 41.24 | 0.023 | −0.158 |
| PCGs | 27.41 | 30.67 | 17.32 | 24.60 | 58.07 | 41.93 | −0.056 | −0.174 |
| cox1 | 26.45 | 32.11 | 18.60 | 22.84 | 58.56 | 41.44 | −0.097 | −0.102 |
| nad4l | 26.26 | 37.71 | 12.79 | 23.23 | 63.97 | 36.03 | −0.179 | −0.290 |
| cox2 | 28.84 | 30.00 | 17.10 | 24.06 | 58.84 | 41.16 | −0.020 | −0.169 |
| atp8 | 33.90 | 29.38 | 15.25 | 21.47 | 63.28 | 36.72 | 0.071 | −0.169 |
| atp6 | 28.51 | 30.70 | 15.20 | 25.58 | 59.21 | 40.79 | −0.037 | −0.255 |
| cox3 | 25.80 | 31.16 | 19.16 | 23.88 | 56.96 | 43.04 | −0.094 | −0.110 |
| nad3 | 22.61 | 32.17 | 15.65 | 29.57 | 54.78 | 45.22 | −0.175 | −0.308 |
| nad4 | 30.26 | 30.26 | 15.79 | 23.68 | 60.53 | 39.47 | 0.000 | −0.200 |
| nad5 | 31.65 | 26.67 | 16.42 | 25.26 | 58.32 | 41.68 | 0.085 | −0.212 |
| nad6 | 14.72 | 42.74 | 26.38 | 16.16 | 57.46 | 42.54 | −0.488 | 0.240 |
| cob | 28.61 | 28.17 | 16.54 | 26.68 | 56.78 | 43.22 | 0.008 | −0.235 |
| nad1 | 23.56 | 30.86 | 19.34 | 26.23 | 54.42 | 45.58 | −0.134 | −0.151 |
| nad2 | 26.17 | 30.75 | 16.14 | 26.93 | 56.92 | 43.08 | −0.080 | −0.251 |
| tRNAs | 32.94 | 29.05 | 19.83 | 18.18 | 61.99 | 38.01 | 0.063 | 0.043 |
| rRNAs | 35.13 | 23.25 | 20.06 | 21.57 | 58.37 | 41.63 | 0.203 | −0.036 |
| Codon | Count | RSCU | Codon | Count | RSCU | Codon | Count | RSCU | Codon | Count | RSCU |
|---|---|---|---|---|---|---|---|---|---|---|---|
| UUU(F) | 189 | 1.01 | UCU(S) | 138 | 1.65 | UAU(Y) | 78 | 0.98 | UGU(C) | 35 | 0.99 |
| UUC(F) | 186 | 0.99 | UCC(S) | 112 | 1.34 | UAC(Y) | 82 | 1.02 | UGC(C) | 36 | 1.01 |
| UUA(L) | 130 | 1.13 | UCA(S) | 80 | 0.95 | UAA(*) | 85 | 1.06 | UGA(W) | 75 | 1.20 |
| UUG(L) | 52 | 0.45 | UCG(S) | 40 | 0.48 | UAG(*) | 76 | 0.94 | UGG(W) | 50 | 0.80 |
| CUU(L) | 158 | 1.37 | CCU(P) | 107 | 1.37 | CAU(H) | 79 | 1.06 | CGU(R) | 26 | 0.89 |
| CUC(L) | 131 | 1.14 | CCC(P) | 88 | 1.13 | CAC(H) | 70 | 0.94 | CGC(R) | 19 | 0.65 |
| CUA(L) | 147 | 1.28 | CCA(P) | 87 | 1.12 | CAA(Q) | 105 | 1.13 | CGA(R) | 40 | 1.37 |
| CUG(L) | 73 | 0.63 | CCG(P) | 30 | 0.38 | CAG(Q) | 81 | 0.87 | CGG(R) | 32 | 1.09 |
| AUU(I) | 130 | 1.03 | ACU(T) | 100 | 1.14 | AAU(N) | 80 | 0.57 | AGU(S) | 48 | 0.57 |
| AUC(I) | 91 | 0.72 | ACC(T) | 102 | 1.17 | AAC(N) | 96 | 0.69 | AGC(S) | 67 | 0.80 |
| AUA(I) | 157 | 1.25 | ACA(T) | 97 | 1.11 | AAA(N) | 242 | 1.74 | AGA(S) | 124 | 1.48 |
| AUG(M) | 95 | 1.00 | ACG(T) | 51 | 0.58 | AAG(K) | 128 | 1.00 | AGG(S) | 62 | 0.74 |
| GUU(V) | 69 | 1.19 | GCU(A) | 85 | 1.31 | GAU(D) | 60 | 0.94 | GGU(G) | 63 | 1.09 |
| GUC(V) | 51 | 0.88 | GCC(A) | 84 | 1.29 | GAC(D) | 67 | 1.06 | GGC(G) | 31 | 0.53 |
| GUA(V) | 73 | 1.26 | GCA(A) | 60 | 0.92 | GAA(E) | 117 | 1.33 | GGA(G) | 89 | 1.53 |
| GUG(V) | 38 | 0.66 | GCG(A) | 31 | 0.48 | GAG(E) | 59 | 0.67 | GGG(G) | 49 | 0.84 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Ma, B.; Li, X.; Lv, Y.; Jiang, X.; Ren, C.; Hu, C.; Luo, P. The Complete Mitochondrial Genome of Stichopus naso (Aspidochirotida: Stichopodidae: Stichopus) and Its Phylogenetic Position. Genes 2022, 13, 825. https://doi.org/10.3390/genes13050825
Li Z, Ma B, Li X, Lv Y, Jiang X, Ren C, Hu C, Luo P. The Complete Mitochondrial Genome of Stichopus naso (Aspidochirotida: Stichopodidae: Stichopus) and Its Phylogenetic Position. Genes. 2022; 13(5):825. https://doi.org/10.3390/genes13050825
Chicago/Turabian StyleLi, Zhuobo, Bo Ma, Xiaomin Li, Ying Lv, Xiao Jiang, Chunhua Ren, Chaoqun Hu, and Peng Luo. 2022. "The Complete Mitochondrial Genome of Stichopus naso (Aspidochirotida: Stichopodidae: Stichopus) and Its Phylogenetic Position" Genes 13, no. 5: 825. https://doi.org/10.3390/genes13050825