
Genome-Wide Association Analysis Reveals Genetic Architecture and Candidate Genes Associated with Grain Yield and Other Traits under Low Soil Nitrogen in Early-Maturing White Quality Protein Maize Inbred Lines


 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Phenotyping
2.3. Phenotypic Data Analysis
2.4. DNA Extraction and Genotyping
2.5. Population Structure and Linkage Disequilibrium (LD)
2.6. Genome-Wide Association Analysis
2.7. Identification of Putative Candidate Genes
3. Results
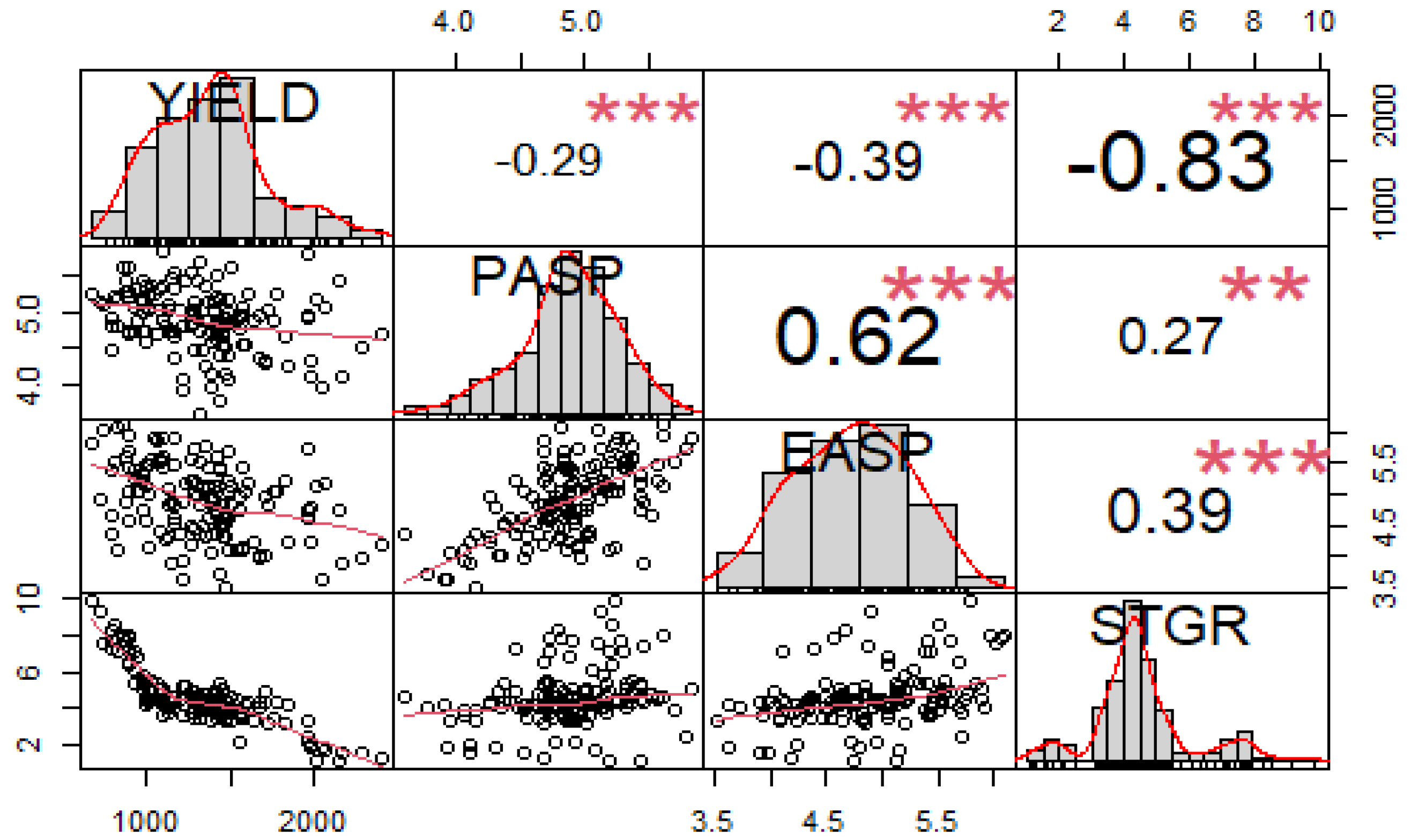
3.1. Evaluation of Phenotypic Traits
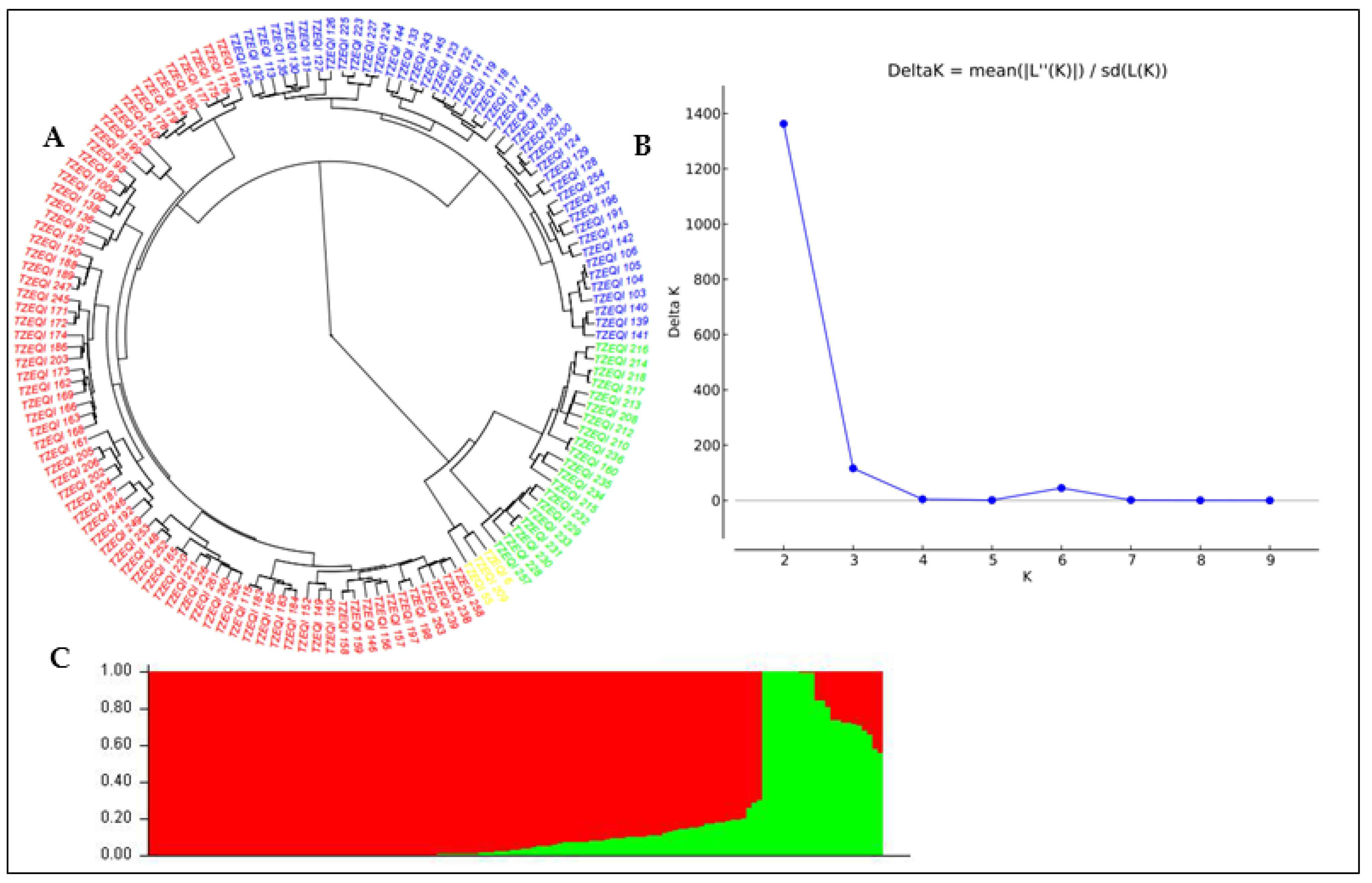
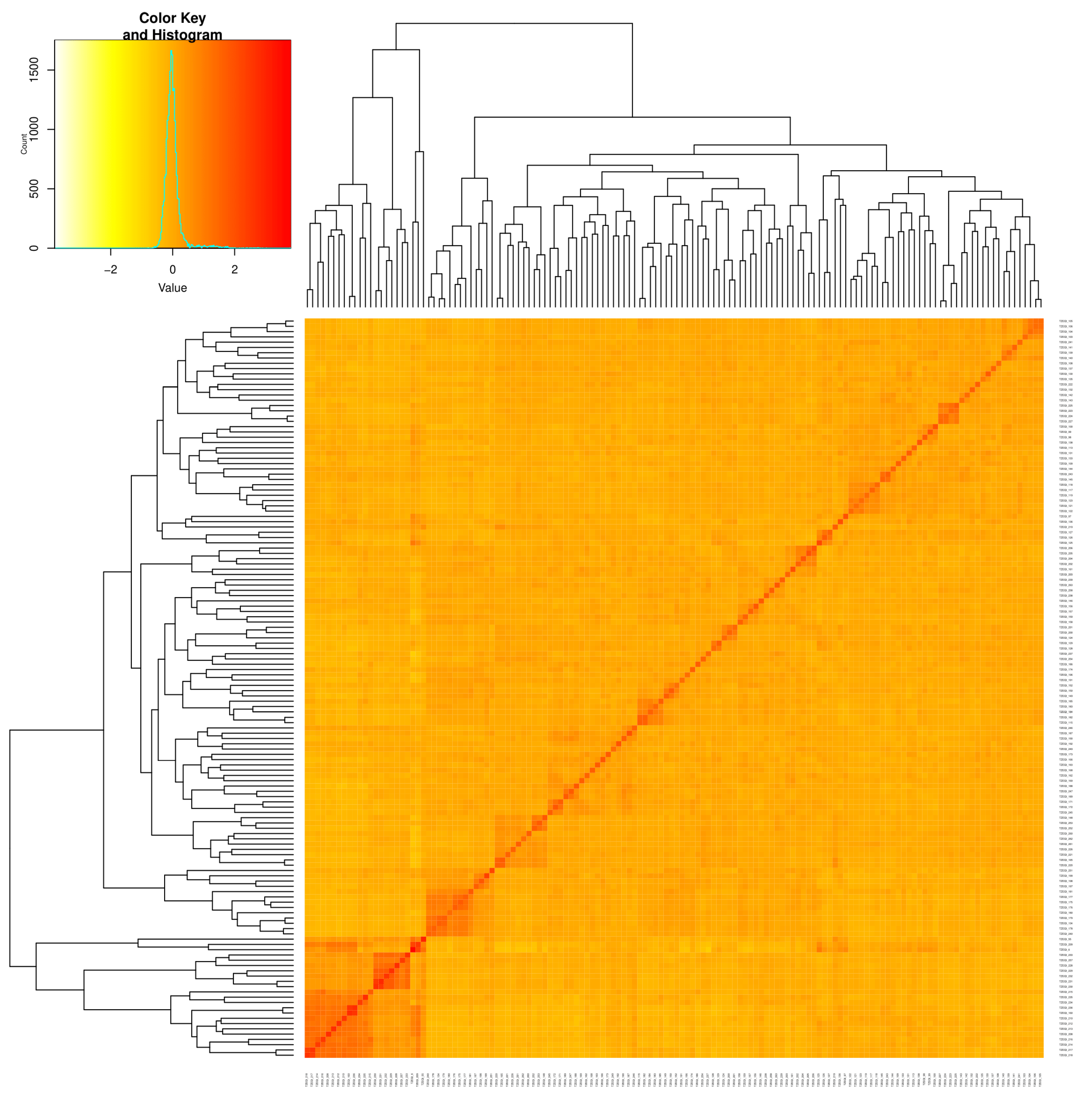
3.2. Population Structure and Genetic Diversity
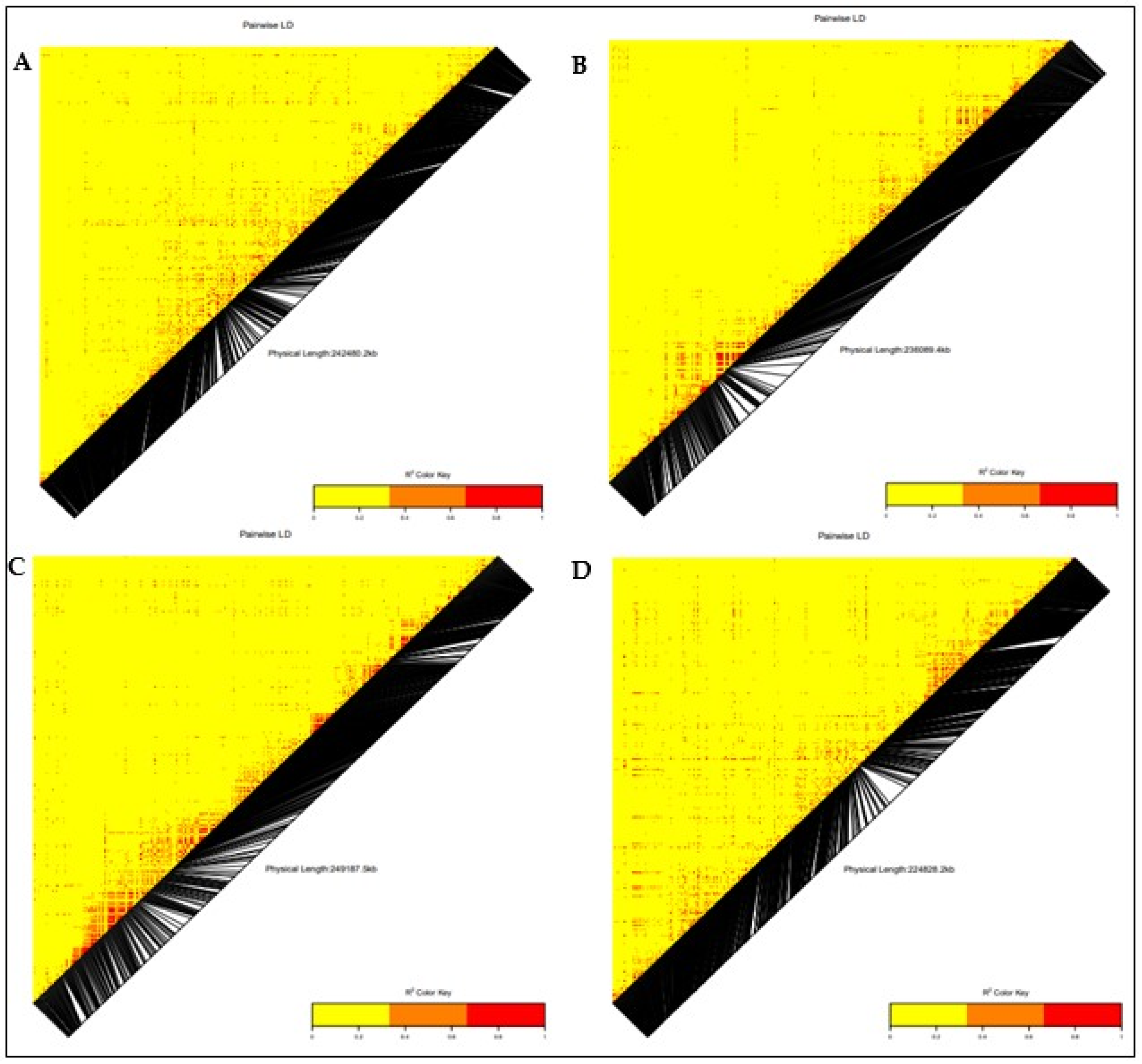
3.3. Linkage Disequilibrium (LD)
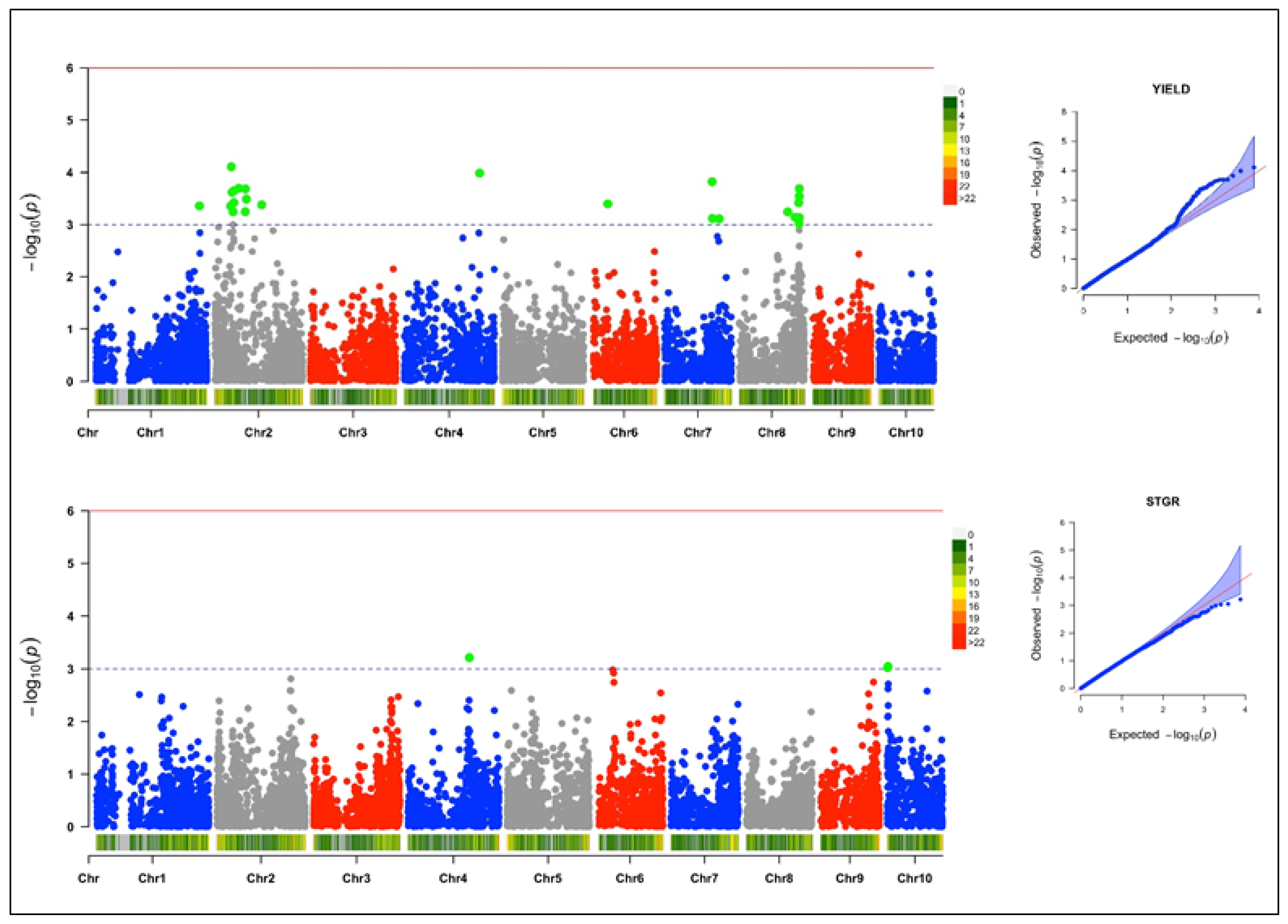
3.4. Genome-Wide Association and LD Analysis
3.4.1. Grain Yield
3.4.2. Stay-Green Characteristic
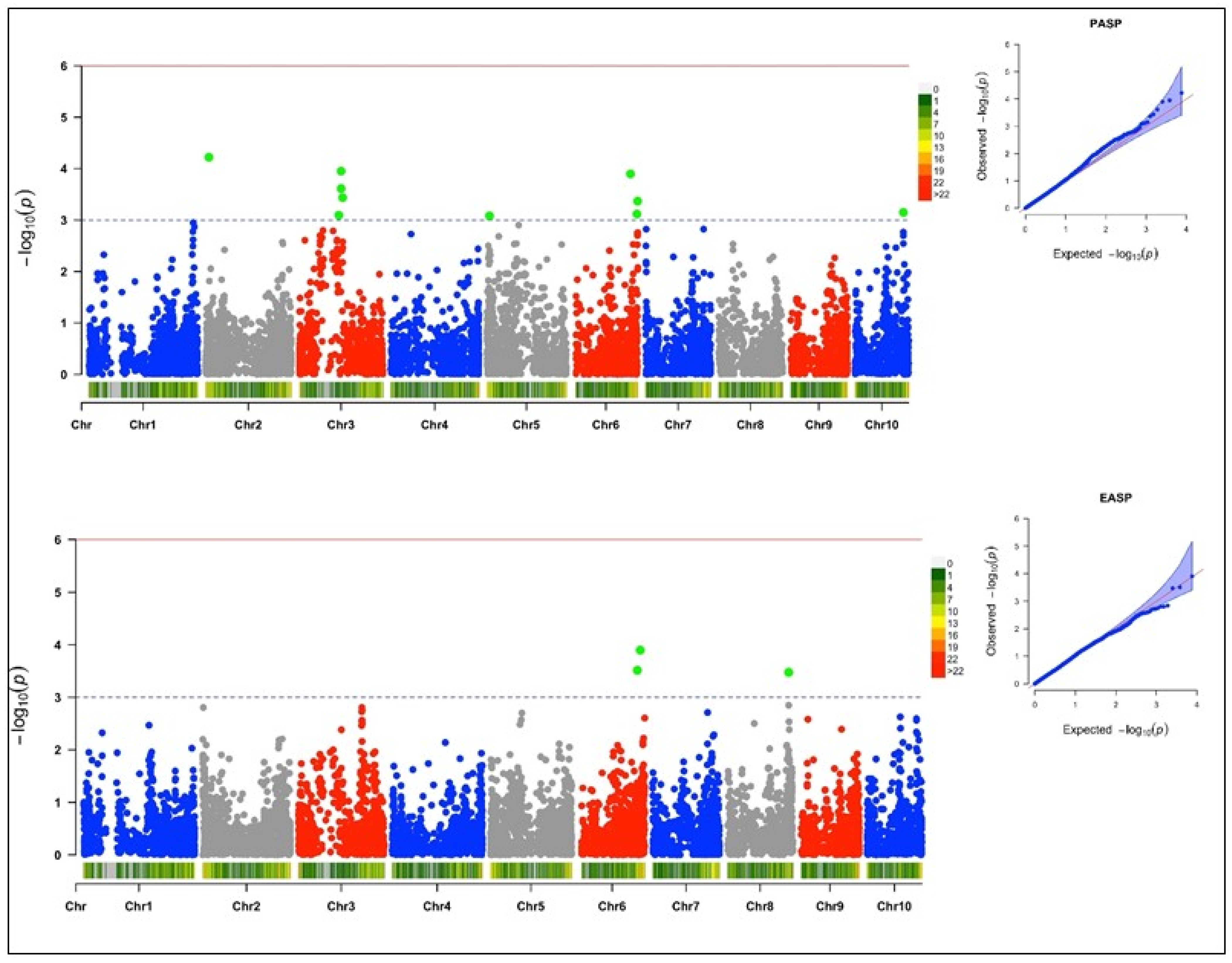
3.4.3. Plant Aspect
3.4.4. Ear Aspect
3.5. Candidate Gene Annotation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hubert, B.; Rosegrant, M.; Van Boekel, M.A.; Ortiz, R. The future of food: Scenarios for 2050. Crop Sci. 2010, 50, S33. [Google Scholar] [CrossRef] [Green Version]
- Shiferaw, B.; Prasanna, B.M.; Hellin, J.; Bänziger, M. Crops that feed the world 6. Past successes and future challenges to the role played by maize in global food security. Food Sec. 2011, 3, 307–327. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Cao, X.H.; Miclăuș, M.; Xu, J.; Xiong, W. The promise of agriculture genomics. Int. Journ. Gen. 2017, 2017, 9743749. [Google Scholar] [CrossRef]
- Perez-de-Castro, A.; Vilanova, S.; Cañizares, J.; Pascual, L.; Blanca, J.; Diez, M.; Prohens, J.; Picó, B. Application of genomic tools in plant breeding. Curr. Gen. 2012, 13, 179–195. [Google Scholar] [CrossRef] [Green Version]
- Tilman, D.; Balzer, C.; Hill, J.; Befort, B.L. Global food demand and the sustainable intensification of agriculture. Proc. Natl. Acad. Sci. USA 2011, 108, 20260–20264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badu-Apraku, B.; Menkir, A.; Ajala, S.; Akinwale, R.; Oyekunle, M.; Obeng-Antwi, K. Performance of tropical early-maturing maize cultivars in multiple stress environments. Can. J. Plant Sci. 2010, 90, 831–852. [Google Scholar] [CrossRef] [Green Version]
- Agrama, H.A.S.; Zakaria, A.G.; Said, F.B.; Tuinstra, M. Identification of quantitative trait loci for nitrogen use efficiency in maize. Mol. Breed. 1999, 5, 187–195. [Google Scholar] [CrossRef]
- Bänziger, M.; Edmeades, G.O.; Beck, D.; Bellon, M. Breeding for Drought and Nitrogen Stress Tolerance in Maize: From Theory to Practice; CIMMYT: El Batán, Mexico, 2000. [Google Scholar]
- Badu-Apraku, B.; Fakorede, M.A.B. Advances in Genetic Enhancement of Early and Extra-Early Maize for Sub-Saharan Africa; Springer: Cham, Switzerland, 2017. [Google Scholar]
- Oikeh, S.O.; Horst, W.J. Agro-physiological responses of tropical maize cultivars to nitrogen fertilization in the moist savanna of West Africa. In Plant Nutrition; Springer: Dordrecht, The Netherlands, 2001; pp. 804–805. [Google Scholar]
- Weber, V.S.; Melchinger, A.E.; Magorokosho, C.; Makumbi, D.; Bänziger, M.; Atlin, G.N. Efficiency of managed stress screening of elite maize hybrids under drought and low nitrogen for yield under rainfed conditions in southern Africa. Crop Sci. 2012, 52, 1011–1020. [Google Scholar] [CrossRef] [Green Version]
- Obeng-Bio, E.; Badu-Apraku, B.; Ifie, B.E.; Danquah, A.; Blay, E.T.; Annor, B. Genetic analysis of grain yield and agronomic traits of early provitamin A quality protein maize inbred lines in contrasting environments. J. Agric. Sci. 2019, 157, 413–433. [Google Scholar] [CrossRef] [Green Version]
- Bhadmus, O.A.; Badu-Apraku, B.; Adeyemo, O.A.; Ogunkanmi, A.L. Genetic Analysis of Early White Quality Protein Maize Inbreds and Derived Hybrids under Low-Nitrogen and Combined Drought and Heat Stress Environments. Plants 2021, 10, 2596. [Google Scholar] [CrossRef]
- Betran, F.J.; Beck, D.; Bänziger, M.; Edmeades, G.O. Genetic analysis of inbred and hybrid grain yield under stress and nonstress environments in tropical maize. Crop Sci. 2003, 43, 807–817. [Google Scholar] [CrossRef]
- Zaidi, P.H.; Srinivasan, G.; Sanchez, C. Relationship between line per se and cross performance under low nitrogen fertility in tropical maize (Zea mays L.). Maydica 2003, 48, 221–231. [Google Scholar]
- Badu-Apraku, B.; Fakorede, M.A.B.; Talabi, A.O.; Oyekunle, M.; Akaogu, I.C.; Akinwale, R.O.; Annor, B.; Melaku, G.; Fasanmade, Y.; Aderounmu, M. Gene action and heterotic groups of early white quality protein maize inbreds under multiple stress environments. Crop Sci. 2015, 56, 183–199. [Google Scholar] [CrossRef] [Green Version]
- Collard, B.C.; Jahufer, M.Z.Z.; Brouwer, J.B.; Pang, E.C.K. An introduction to markers, quantitative trait loci (QTL) mapping and marker-assisted selection for crop improvement: The basic concepts. Euphytica 2005, 142, 169–196. [Google Scholar] [CrossRef]
- He, J.; Zhao, X.; Laroche, A.; Lu, Z.X.; Liu, H.; Li, Z. Genotyping-by-sequencing (GBS), an ultimate marker-assisted selection (MAS) tool to accelerate plant breeding. Front. Plant Sci. 2014, 5, 484. [Google Scholar] [CrossRef] [Green Version]
- Farfan, I.D.B.; De La Fuente, G.N.; Murray, S.C.; Isakeit, T.; Huang, P.C.; Warburton, M.; Kolomiets, M. Genome wide association study for drought, aflatoxin resistance, and important agronomic traits of maize hybrids in the sub-tropics. PLoS ONE 2015, 10, e0117737. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Shrestha, R.; Ding, J.; Zheng, H.; Mu, C.; Wu, J.; Mahuku, G. Genome-wide association study and QTL mapping reveal genomic loci associated with Fusarium ear rot resistance in tropical maize germplasm. G3 Genes Genomes Genet. 2016, 6, 3803–3815. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Cairns, J.E.; Babu, R.; Gowda, M.; Makumbi, D.; Magorokosho, C.; Zhang, A.; Liu, Y.; Wang, N.; Hao, Z.; et al. Genome-Wide Association Mapping and Genomic Prediction Analyses Reveal the Genetic Architecture of Grain Yield and Flowering Time Under Drought and Heat Stress Conditions in Maize. Plant Sci. 2019, 9, 1919. [Google Scholar] [CrossRef] [Green Version]
- Adewale, S.A.; Badu-Apraku, B.; Akinwale, R.O.; Paterne, A.A.; Gedil, M.; Garcia-Oliveira, A.L. Genome-wide association study of Striga resistance in early maturing white tropical maize inbred lines. BMC Plant Biol. 2020, 20, 203. [Google Scholar] [CrossRef]
- Longmei, N.; Gill, G.K.; Zaidi, P.H.; Kumar, R.; Nair, S.K.; Hindu, V.; Vikal, Y. Genome wide association mapping for heat tolerance in sub-tropical maize. BMC Genom. 2021, 22, 154. [Google Scholar] [CrossRef]
- López-Malvar, A.; Butron, A.; Malvar, R.A.; McQueen-Mason, S.J.; Faas, L.; Gómez, L.D.; Santiago, R. Association mapping for maize stover yield and saccharification efficiency using a multiparent advanced generation intercross (MAGIC) population. Sci. Rep. 2021, 11, 3425. [Google Scholar] [CrossRef] [PubMed]
- Ertiro, B.T.; Labuschagne, M.; Olsen, M.; Das, B.; Prasanna, B.M.; Gowda, M. Genetic dissection of nitrogen use efficiency in tropical maize through genome-wide association and genomic prediction. Front. Plant Sci. 2020, 11, 474. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Xu, S.; Zhang, X.; Li, Y.; Chang, L.; Wang, Y.; Xue, J. Mining of candidate genes for nitrogen use efficiency in maize based on genome-wide association study. Mol. Breed. 2020, 40, 83. [Google Scholar] [CrossRef]
- Badu-Apraku, B.; Fakorede, M.A.B.; Oyekunle, M.; Akinwale, R.O. Selection of extra-early maize inbreds under low N and drought at flowering and grain-filling for hybrid production. Maydica 2011, 56, 1721–1735. [Google Scholar]
- Badu-Apraku, B.; Akinwale, R.O.; Franco, J.; Oyekunle, M. Assessment of reliability of secondary traits in selecting for improved grain yield in drought and low-nitrogen environments. Crop Sci. 2012, 52, 2050–2062. [Google Scholar] [CrossRef]
- Ifie, B.E.; Badu-Apraku, B.; Gracen, V.; Danquah, E.Y. Genetic analysis of grain yield of IITA and CIMMYT early maize inbreds under Striga-infested and low-soil-nitrogen environments. Crop Sci. 2015, 55, 610–623. [Google Scholar] [CrossRef]
- Talabi, A.O.; Badu-Apraku, B.; Fakorede, M.A.B. Genetic Variances and Relationship among Traits of an Early Maturing Maize Population under Drought-stress and Low Nitrogen Environments. Crop Sci. 2017, 57, 681–692. [Google Scholar] [CrossRef] [Green Version]
- Annor, B.; Badu-Apraku, B. Gene action controlling grain yield and other agronomic traits of extra-early quality protein maize under stress and non-stress conditions. Euphytica 2016, 212, 213–228. [Google Scholar] [CrossRef]
- Oyekale, S.A.; Badu-Apraku, B.; Adetimirin, V.O. Combining ability of extra-early biofortified maize inbreds under Striga infestation and low soil nitrogen. Crop Sci. 2020, 60, 1925–1934. [Google Scholar] [CrossRef]
- SAS Institute Inc. SAS User’s Guide: Statistics; Version 9.4; SAS Institute Inc.: Cary, NC, USA, 2017. [Google Scholar]
- Alvarado, G.; Rodríguez, F.M.; Pacheco, A.; Burgueño, J.; Crossa, J.; Vargas, M.; Lopez-Cruz, M.A. META-R: A software to analyze data from multi-environment plant breeding trials. Crop J. 2020, 8, 745–756. [Google Scholar] [CrossRef]
- Peterson, B.G.; Carl, P.; Boudt, K.; Bennett, R.; Ulrich, J.; Zivot, E.; Cornilly, D.; Hung, E.; Lestel, M.; Balkissoon, K.; et al. Package ‘Performanceanalytics’. R Team Cooperation. Available online: https://cran.r-project.org/web/packages/PerformanceAnalytics/PerformanceAnalytics.pdf (accessed on 27 March 2022).
- Hallauer, A.R.; Miranda, J.B. Quantitative Genetics in Maize Breeding, 2nd ed.; Iowa State University Press: Ames, IA, USA, 1988. [Google Scholar]
- Azmach, G.; Gedil, M.; Menkir, A.; Spillane, C. Marker-trait association analysis of functional gene markers for provitamin A levels across diverse tropical yellow maize inbred lines. BMC Plant Biol. 2013, 13, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaccoud, D.; Peng, K.; Feinstein, D.; Kilian, A. Diversity arrays: A solid state technology for sequence information independent genotyping. Nucleic Acids Res. 2001, 29, e25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, Y.; Peluso, P.; Shi, J.; Tiffany, L.; Stitzer, M.C.; Wang, B. Improved maize reference genome with single-molecule technologies. Nature 2017, 546, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2010, 155, 945–959. [Google Scholar] [CrossRef]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Earl, D.A.; vonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing Structure output and implementing the Evanno method. Conserv. Genet. Resour. 2013, 4, 359. [Google Scholar] [CrossRef]
- Paradis, E.; Schliep, K. Ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Sham, P.C. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Lipka, A.E.; Tian, F.; Wang, Q.; Peiffer, J.; Li, M.; Bradbury, P.J.; Gore, M.A.; Buckler, E.S.; Zhang, Z. GAPIT: Genome association and prediction integrated tool. Bioinformatics 2012, 28, 2397–2399. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Bi, I.V.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.D. QQman: An R package for visualizing GWAS results using QQ and Manhattan plots. Biorxiv 2014, 2014, 005165. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Burghardt, L.T.; Young, N.D.; Tiffin, P. A guide to genome-wide association mapping in plants. Curr. Protoc. Plant Biol. 2017, 2, 22–38. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Blay, S.; McNeney, B.; Graham, J. LDheatmap: An R function for graphical display of pairwise linkage disequilibria between single nucleotide polymorphisms. J. Stat. Softw. 2006, 16, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Paterne, A.A.; Norman, P.E.; Asiedu, R.; Asfaw, A. Identification of quantitative trait nucleotides and candidate genes for tuber yield and mosaic virus tolerance in an elite population of white guinea yam (Dioscorea rotundata) using genome-wide association scan. BMC Plant Biol. 2021, 21, 552. [Google Scholar] [CrossRef]
- Eltaher, S.; Sallam, A.; Belamkar, V.; Emara, H.A.; Nower, A.A.; Salem, K.F.; Baenziger, P.S. Genetic diversity and population structure of F3: 6 Nebraska winter wheat genotypes using genotyping-by-sequencing. Front. Genet. 2018, 9, 76. [Google Scholar] [CrossRef]
- Soto-Cerda, B.J.; Cloutier, S. Association mapping in plant genomes. Genet. Divers. Plants 2012, 29–54. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, N.; Armbruster, U.; Iven, T.; Brückle, L.; Melzer, M.; Feussner, I.; Jahns, P. Tissue-specific accumulation and regulation of zeaxanthin epoxidase in Arabidopsis reflect the multiple functions of the enzyme in plastids. Plant Cell Physiol. 2015, 56, 346–357. [Google Scholar] [CrossRef] [Green Version]
- Park, H.Y.; Seok, H.Y.; Park, B.K.; Kim, S.H.; Goh, C.H.; Lee, B.H.; Moon, Y.H. Overexpression of Arabidopsis ZEP enhances tolerance to osmotic stress. Biochem. Biophys. Res. Commun. 2008, 375, 80–85. [Google Scholar] [CrossRef]
- Chen, K.; Li, G.J.; Bressan, R.A.; Song, C.P.; Zhu, J.K.; Zhao, Y. Abscisic acid dynamics, signaling, and functions in plants. J. Integr. Plant Biol. 2020, 62, 25–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demmig-Adams, B.; Adams, W.W., III. The role of xanthophyll cycle carotenoids in the protection of photosynthesis. Trends Plant Sci. 1996, 1, 21–26. [Google Scholar] [CrossRef]
- Latowski, D.; Kuczyńska, P.; Strzałka, K. Xanthophyll cycle–a mechanism protecting plants against oxidative stress. Redox Rep. 2011, 16, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Cecchetti, V.; Altamura, M.M.; Falasca, G.; Costantino, P.; Cardarelli, M. Auxin regulates Arabidopsis anther dehiscence, pollen maturation, and filament elongation. Plant Cell 2008, 20, 1760–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal, E.A.; Araus, V.; Lu, C.; Parry, G.; Green, P.J.; Coruzzi, G.M.; Gutiérrez, R.A. Nitrate-responsive miR393/AFB3 regulatory module controls root system architecture in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2010, 107, 4477–4482. [Google Scholar] [CrossRef] [Green Version]
- Schwechheimer, C.; Isono, E. The COP9 signalosome and its role in plant development. Euro. J. Cell Biol. 2010, 89, 157–162. [Google Scholar] [CrossRef]
- Shang, Y.; Wang, K.; Sun, S.; Zhou, J.; Yu, J.Q. COP9 Signalosome CSN4 and CSN5 subunits are involved in Jasmonate-dependent defense against root-knot nematode in tomato. Front. Plant Sci. 2019, 10, 1223–1233. [Google Scholar] [CrossRef]
- Chen, C.N.; Chu, C.C.; Zentella, R.; Pan, S.M.; Ho, T.H.D. AtHVA22 gene family in Arabidopsis: Phylogenetic relationship, ABA and stress regulation, and tissue-specific expression. Plant Mol. Biol. 2002, 49, 631–642. [Google Scholar] [CrossRef]
- Tan, J.; Wu, F.; Wan, J. Flowering time regulation by the CONSTANS-Like gene OsCOL10. Plant Signal. Behav. 2017, 12, 798–812. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Zhang, Y.; Zhang, M.; Zhan, X.; Shen, X.; Yu, P.; Cao, L. The rice CONSTANS-like protein OsCOL15 suppresses flowering by promoting Ghd7 and repressing RID1. Biochem. Biophys. Res. Commun. 2018, 495, 1349–1355. [Google Scholar] [CrossRef]
- Kim, M.C.; Lee, S.H.; Kim, J.K.; Chun, H.J.; Choi, M.S.; Chung, W.S.; Cho, M.J. Mlo, a modulator of plant defense and cell death, is a novel calmodulin-binding protein: Isolation and characterization of a rice Mlo homologue. J. Biol. Chem. 2002, 277, 19304–19314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y. The Role of SNF1-Related Protein Kinase 1 (SnRK1) in Regulating Intermediary Metabolism in Arabidopsis Thaliana. Ph.D. Thesis, University of Guelph, Guelph, ON, Canada, 2017. Available online: http://hdl.handle.net/10214/11945 (accessed on 14 September 2021).
- Waqas, M.; Feng, S.; Amjad, H.; Letuma, P.; Zhan, W.; Li, Z.; Lin, W. Protein phosphatase (PP2C9) induces protein expression differentially to mediate nitrogen utilization efficiency in rice under nitrogen-deficient condition. Int. J. Mol. Sci. 2018, 19, 2827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stegmann, M.; Anderson, R.G.; Westphal, L.; Rosahl, S.; McDowell, J.M.; Trujillo, M. The exocyst subunit Exo70B1 is involved in the immune response of Arabidopsis thaliana to different pathogens and cell death. Plant Signal. Behav. 2013, 8, e27421. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Liu, N.; Gao, C.; Rui, L.; Tang, D. The Pseudomonas syringae effector AvrPtoB associates with and ubiquitinates Arabidopsis exocyst subunit EXO70B1. Front. Plant Sci. 2019, 10, 1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, G.; Ma, Z.; Zhang, L.; Xing, S.; Hou, X.; Deng, J.; Gu, H. Arabidopsis AtBECLIN 1/AtAtg6/AtVps30 is essential for pollen germination and plant development. Cell Res. 2007, 17, 249–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coe, J.; Kupitz, C.; Basu, S.; Conrad, C.E.; Roy-Chowdhury, S.; Fromme, R.; Fromme, P. Crystallization of photosystem II for time-resolved structural studies using an X-ray free electron laser. Methods Enzymol. 2015, 557, 459–482. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Maekawa, S.; Yasuda, S.; Sonoda, Y.; Katoh, E.; Ichikawa, T.; Yamaguchi, J. CNI1/ATL31, a RING-type ubiquitin ligase that functions in the carbon/nitrogen response for growth phase transition in Arabidopsis seedlings. Plant J. 2009, 60, 852–864. [Google Scholar] [CrossRef]
- Jadid, N.; Mialoundama, A.S.; Heintz, D.; Ayoub, D.; Erhardt, M.; Mutterer, J.; Bouvier, F. DOLICHOL PHOSPHATE MANNOSE SYNTHASE1 mediates the biogenesis of isoprenyl-linked glycans and influences development, stress response, and ammonium hypersensitivity in Arabidopsis. Plant Cell 2011, 23, 1985–2005. [Google Scholar] [CrossRef] [Green Version]
- Cui, H.; Kong, D.; Liu, X.; Hao, Y. SCARECROW, SCR-LIKE 23 and SHORT-ROOT control bundle sheath cell fate and function in Arabidopsis thaliana. Plant J. 2014, 78, 319–327. [Google Scholar] [CrossRef]
- Césari, S.; Kanzaki, H.; Fujiwara, T.; Bernoux, M.; Chalvon, V.; Kawano, Y.; Kroj, T. The NB-LRR proteins RGA 4 and RGA 5 interact functionally and physically to confer disease resistance. EMBO J. 2014, 33, 1941–1959. [Google Scholar] [CrossRef]
- Sekhwal, M.K.; Li, P.; Lam, I.; Wang, X.; Cloutier, S.; You, F.M. Disease resistance gene analogs (RGAs) in plants. Int. J. Mol. Sci. 2015, 16, 19248–19290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koops, P.; Pelser, S.; Ignatz, M.; Klose, C.; Marrocco-Selden, K.; Kretsch, T. EDL3 is an F-box protein involved in the regulation of abscisic acid signalling in Arabidopsis thaliana. J. Exp. Bot. 2011, 62, 5547–5560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, D.; Yang, H.; Venglat, P.; Cao, Y.; Wen, R.; Ren, M.; Datla, R. POPCORN functions in the auxin pathway to regulate embryonic body plan and meristem organization in Arabidopsis. Plant Cell 2011, 23, 4348–4367. [Google Scholar] [CrossRef] [Green Version]
- Richter, R.; Behringer, C.; Müller, I.K.; Schwechheimer, C. The GATA-type transcription factors GNC and GNL/CGA1 repress gibberellin signaling downstream from DELLA proteins and PHYTOCHROME-INTERACTING FACTORS. Genes Dev. 2010, 24, 2093–2104. Available online: http://www.genesdev.org/cgi/doi/10.1101/gad.594910 (accessed on 14 September 2021). [CrossRef] [PubMed] [Green Version]
- Hudson, D.; Guevara, D.; Yaish, M.W.; Hannam, C.; Long, N.; Clarke, J.D.; Rothstein, S.J. GNC and CGA1 modulate chlorophyll biosynthesis and glutamate synthase (GLU1/Fd-GOGAT) expression in Arabidopsis. PLoS ONE 2011, 6, e26765. [Google Scholar] [CrossRef]
- Lancien, M.; Martin, M.; Hsieh, M.H.; Leustek, T.; Goodman, H.; Coruzzi, G.M. Arabidopsis glt1-T mutant defines a role for NADH-GOGAT in the non-photorespiratory ammonium assimilatory pathway. Plant J. 2002, 29, 347–358. [Google Scholar] [CrossRef]
- Tamura, W.; Hidaka, Y.; Tabuchi, M.; Kojima, S.; Hayakawa, T.; Sato, T.; Yamaya, T. Reverse genetics approach to characterize a function of NADH-glutamate synthase1 in rice plants. Amino Acids 2010, 39, 1003–1012. [Google Scholar] [CrossRef]
- Morosini, J.S.; Mendonca, L.D.F.; Lyra, D.H.; Galli, G.; Vidotti, M.S.; Fritsche-Neto, R. Association mapping for traits related to nitrogen use efficiency in tropical maize lines under field conditions. Plant Soil 2017, 421, 453–463. [Google Scholar] [CrossRef]
- Hu, Y.; Xie, Q.; Chua, N.H. The Arabidopsis auxin-inducible gene ARGOS controls lateral organ size. Plant Cell 2003, 15, 1951–1961. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Sang, Y.; Song, J.; Gao, X.Q.; Zhang, X. Expression of a rice OsARGOS gene in Arabidopsis promotes cell division and expansion and increases organ size. J. Genet. Genom. 2009, 36, 31–40. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SV | DF | YIELD | PASP | EASP | STGR |
|---|---|---|---|---|---|
| ENV | 1 | 28,069,163.12 ** | 116.81 ** | 130.65 ** | 276.07 ** |
| Block (ENV*Rep) | 48 | 519,164.92 ** | 1.69 ** | 2.66 ** | 2.27 ** |
| Rep (ENV) | 2 | 314,250.52 | 8.35 ** | 11.94 ** | 16.55 ** |
| Genotypes | 168 | 438,971.11 ** | 0.70 ** | 1.28 ** | 2.23 ** |
| ENV*Genotypes | 168 | 456,508.9 ** | 0.47 | 0.62 | 1.31 |
| Error | 288 | 241,768.5 | 0.41 | 0.60 | 1.07 |
| H2 | 0.57 | 0.41 | 0.45 | 0.43 |
| Traits | SNP | Chr | Position | p-Value | MAF | Marker R2 | LOD |
|---|---|---|---|---|---|---|---|
| Grain yield | S2_46273057 | 2 | 46273057 | 7.8 × 10−5 | 0.05 | 0.17 | 4.11 |
| S4_209096186 | 4 | 209096186 | 1.0 × 10−4 | 0.20 | 0.17 | 3.99 | |
| S7_131622616 | 7 | 131622616 | 1.5 × 10−4 | 0.08 | 0.16 | 3.82 | |
| S2_67297792 | 2 | 67297792 | 2.0 × 10−4 | 0.31 | 0.16 | 3.69 | |
| S8_166330750 | 8 | 166330750 | 2.1 × 10−4 | 0.29 | 0.16 | 3.69 | |
| S2_85668156 | 2 | 85668156 | 2.1 × 10−4 | 0.04 | 0.16 | 3.68 | |
| S2_54250098 | 2 | 54250098 | 2.2 × 10−4 | 0.12 | 0.16 | 3.65 | |
| S2_48447873 | 2 | 48447873 | 2.4 × 10−4 | 0.05 | 0.16 | 3.62 | |
| S8_166202087 | 8 | 166202087 | 2.9 × 10−4 | 0.29 | 0.16 | 3.54 | |
| S8_166330684 | 8 | 166330684 | 2.9 × 10−4 | 0.28 | 0.16 | 3.54 | |
| S2_88084334 | 2 | 88084334 | 3.3 × 10−4 | 0.06 | 0.15 | 3.49 | |
| S2_53491979 | 2 | 53491979 | 3.8 × 10−4 | 0.06 | 0.15 | 3.42 | |
| S8_164481914 | 8 | 164481914 | 3.8 × 10−4 | 0.21 | 0.15 | 3.42 | |
| S6_45087496 | 6 | 45087496 | 4.0 × 10−4 | 0.02 | 0.15 | 3.40 | |
| S2_130453666 | 2 | 130453666 | 4.2 × 10−4 | 0.04 | 0.15 | 3.38 | |
| S1_287891383 | 1 | 287891383 | 4.4 × 10−4 | 0.37 | 0.15 | 3.36 | |
| S2_44598915 | 2 | 44598915 | 4.4 × 10−4 | 0.07 | 0.15 | 3.36 | |
| S2_51434022 | 2 | 51434022 | 5.7 × 10−4 | 0.04 | 0.15 | 3.24 | |
| S2_85053187 | 2 | 85053187 | 5.7 × 10−4 | 0.04 | 0.15 | 3.24 | |
| S8_134334368 | 8 | 134334368 | 5.7 × 10−4 | 0.05 | 0.15 | 3.24 | |
| S8_154518069 | 8 | 154518069 | 7.3 × 10−4 | 0.14 | 0.14 | 3.13 | |
| S8_166462404 | 8 | 166462404 | 7.4 × 10−4 | 0.30 | 0.14 | 3.13 | |
| S7_132400339 | 7 | 132400339 | 7.6 × 10−4 | 0.08 | 0.14 | 3.12 | |
| S7_151236665 | 7 | 151236665 | 7.8 × 10−4 | 0.19 | 0.14 | 3.11 | |
| Stay green characteristic | S4_168465704 | 4 | 168465704 | 6.1 × 10−4 | 0.24 | 0.09 | 3.21 |
| S10_2268677 | 10 | 2268677 | 8.9 × 10−4 | 0.24 | 0.08 | 3.05 | |
| S10_1417870 | 10 | 1417870 | 9.4 × 10−4 | 0.50 | 0.08 | 3.02 | |
| Plant aspect | S2_10153860 | 2 | 10153860 | 6.0 × 10−5 | 0.16 | 0.16 | 4.22 |
| S3_119115762 | 3 | 119115762 | 1.1 × 10−4 | 0.10 | 0.15 | 3.95 | |
| S6_159734917 | 6 | 159734917 | 1.2 × 10−4 | 0.19 | 0.15 | 3.90 | |
| S3_119024277 | 3 | 119024277 | 2.4 × 10−4 | 0.12 | 0.14 | 3.61 | |
| S3_123440369 | 3 | 123440369 | 3.7 × 10−4 | 0.10 | 0.13 | 3.44 | |
| S6_179917351 | 6 | 179917351 | 4.3 × 10−4 | 0.09 | 0.13 | 3.37 | |
| S10_136641842 | 10 | 136641842 | 7.1 × 10−4 | 0.34 | 0.12 | 3.15 | |
| S6_177721271 | 6 | 177721271 | 7.6 × 10−4 | 0.15 | 0.12 | 3.12 | |
| S3_112551813 | 3 | 112551813 | 8.0 × 10−4 | 0.09 | 0.12 | 3.10 | |
| S5_8518748 | 5 | 8518748 | 8.3 × 10−4 | 0.10 | 0.12 | 3.08 | |
| Ear aspect | S6_167701917 | 6 | 167701917 | 1.3 × 10−4 | 0.06 | 0.17 | 3.90 |
| S6_159734917 | 6 | 159734917 | 3.1 × 10−4 | 0.19 | 0.16 | 3.51 | |
| S8_169668528 | 8 | 169668528 | 3.3 × 10−4 | 0.11 | 0.16 | 3.48 |
| Traits | Chr | Position | Gene ID | Encoding Products | Functions |
|---|---|---|---|---|---|
| Grain yield | 2 | 46273057 | GRMZM2G127139 | Zeaxanthin epoxidase | Biosynthesis of abscisic acid |
| GRMZM2G015610 | Protein phosphatase | Unknown function | |||
| 1 | 287891383 | GRMZM2G067964 | Dolichol-phosphate mannosyltransferase | Plant growth and development | |
| 2 | 67297792 | GRMZM5G848945 | Protein AUXIN SIGNALING F-BOX | Primary and lateral root development | |
| GRMZM5G898735 | high chlorophyll fluorescence 106 | Photosynthesis | |||
| 2 | 85668156 | GRMZM2G049141 | E3 ubiquitin-protein ligase UPL3 | Unknown function | |
| 2 | 54250098 | GRMZM2G107588 | COP9 signalosome complex subunit 8 | Plant growth and stress tolerance | |
| GRMZM2G157822 | HVA22-like protein f | Plant reproductive development | |||
| 2 | 48447873 | GRMZM2G106108 | zinc finger protein CONSTANS-LIKE 1 | Plant flowering time | |
| GRMZM2G028543 | putative RING zinc finger domain superfamily protein | Plant growth and development | |||
| GRMZM2G031331 | mlo defense gene homolog 3 | Plant disease resistance and leaf cell death | |||
| 2 | 88084334 | GRMZM2G012942 | SNARE-interacting protein KEULE | ||
| 7 | 132400339 | GRMZM2G082653 | E3 ubiquitin-protein ligase ATL31 | Cellular response to nitrogen levels | |
| 8 | 166330750 | GRMZM2G003493 | SNF1-related protein kinase regulatory subunit γ-1 | Assimilation of nitrogen in plants | |
| GRMZM2G003518 | exocyst complex component EXO70B1 | Plant defense response to stress | |||
| GRMZM2G027857 | beclin-1-like protein | Cellular response to nitrogen | |||
| 8 | 164481914 | GRMZM2G005290 | chitinase CLP | Root and shoot development | |
| GRMZM2G005433 | photosystem II reaction center PSB28 protein | Photosynthesis in plants | |||
| Stay Green Characteristic | 10 | 1417870 | AC198366.3_FG004 | Scarecrow-like protein 3 | Plant leaf development |
| GRMZM2G180254 | disease resistance protein RGA5 | Plant cell death | |||
| Plant Aspect | 6 | 159734917 | GRMZM2G440849 | putative disease resistance protein RGA3 | Disease resistance in plant |
| GRMZM2G440968 | cystatin 3 | Unknown function | |||
| GRMZM2G389301 | EID1-like F-box protein 3 | Plant growth and development | |||
| 10 | 136641842 | GRMZM2G169645 | putative RING zinc finger domain superfamily protein | Unknown function | |
| 5 | 8518748 | GRMZM2G401040 | ATP synthase F1, delta subunit family protein | Energy conversion in photosynthesis | |
| GRMZM2G065822 | WD repeat-containing protein PCN | Leaf formation and development | |||
| GRMZM2G065896 | GATA transcription factor 25 | Chlorophyll biosynthesis | |||
| Ear Aspect | 6 | 167701917 | GRMZM2G375064 | glutamate synthase 2 (NADH), chloroplastic | Nitrogen metabolism |
| 6 | 159734917 | GRMZM2G440849 | putative disease resistance protein RGA3 | Plant resistance to diseases | |
| GRMZM2G440968 | cystatin 3 | Unknown function | |||
| GRMZM2G389301 | EID1-like F-box protein 3 | Regulates plant growth and development | |||
| 8 | 169668528 | GRMZM2G171996 | protein auxin-regulated gene involved in organ size | Plant organ development |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhadmus, O.A.; Badu-Apraku, B.; Adeyemo, O.A.; Agre, P.A.; Queen, O.N.; Ogunkanmi, A.L. Genome-Wide Association Analysis Reveals Genetic Architecture and Candidate Genes Associated with Grain Yield and Other Traits under Low Soil Nitrogen in Early-Maturing White Quality Protein Maize Inbred Lines. Genes 2022, 13, 826. https://doi.org/10.3390/genes13050826
Bhadmus OA, Badu-Apraku B, Adeyemo OA, Agre PA, Queen ON, Ogunkanmi AL. Genome-Wide Association Analysis Reveals Genetic Architecture and Candidate Genes Associated with Grain Yield and Other Traits under Low Soil Nitrogen in Early-Maturing White Quality Protein Maize Inbred Lines. Genes. 2022; 13(5):826. https://doi.org/10.3390/genes13050826
Chicago/Turabian StyleBhadmus, Olatunde A., Baffour Badu-Apraku, Oyenike A. Adeyemo, Paterne A. Agre, Offornedo N. Queen, and Adebayo L. Ogunkanmi. 2022. "Genome-Wide Association Analysis Reveals Genetic Architecture and Candidate Genes Associated with Grain Yield and Other Traits under Low Soil Nitrogen in Early-Maturing White Quality Protein Maize Inbred Lines" Genes 13, no. 5: 826. https://doi.org/10.3390/genes13050826
APA StyleBhadmus, O. A., Badu-Apraku, B., Adeyemo, O. A., Agre, P. A., Queen, O. N., & Ogunkanmi, A. L. (2022). Genome-Wide Association Analysis Reveals Genetic Architecture and Candidate Genes Associated with Grain Yield and Other Traits under Low Soil Nitrogen in Early-Maturing White Quality Protein Maize Inbred Lines. Genes, 13(5), 826. https://doi.org/10.3390/genes13050826