
The Use of DArTseq Technology to Identify New SNP and SilicoDArT Markers Related to the Yield-Related Traits Components in Maize

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Results
2.1. Analysis of the Size of the Components of the Yield of Inbred Maize Lines
2.1.1. Multi-Traits Comparisons
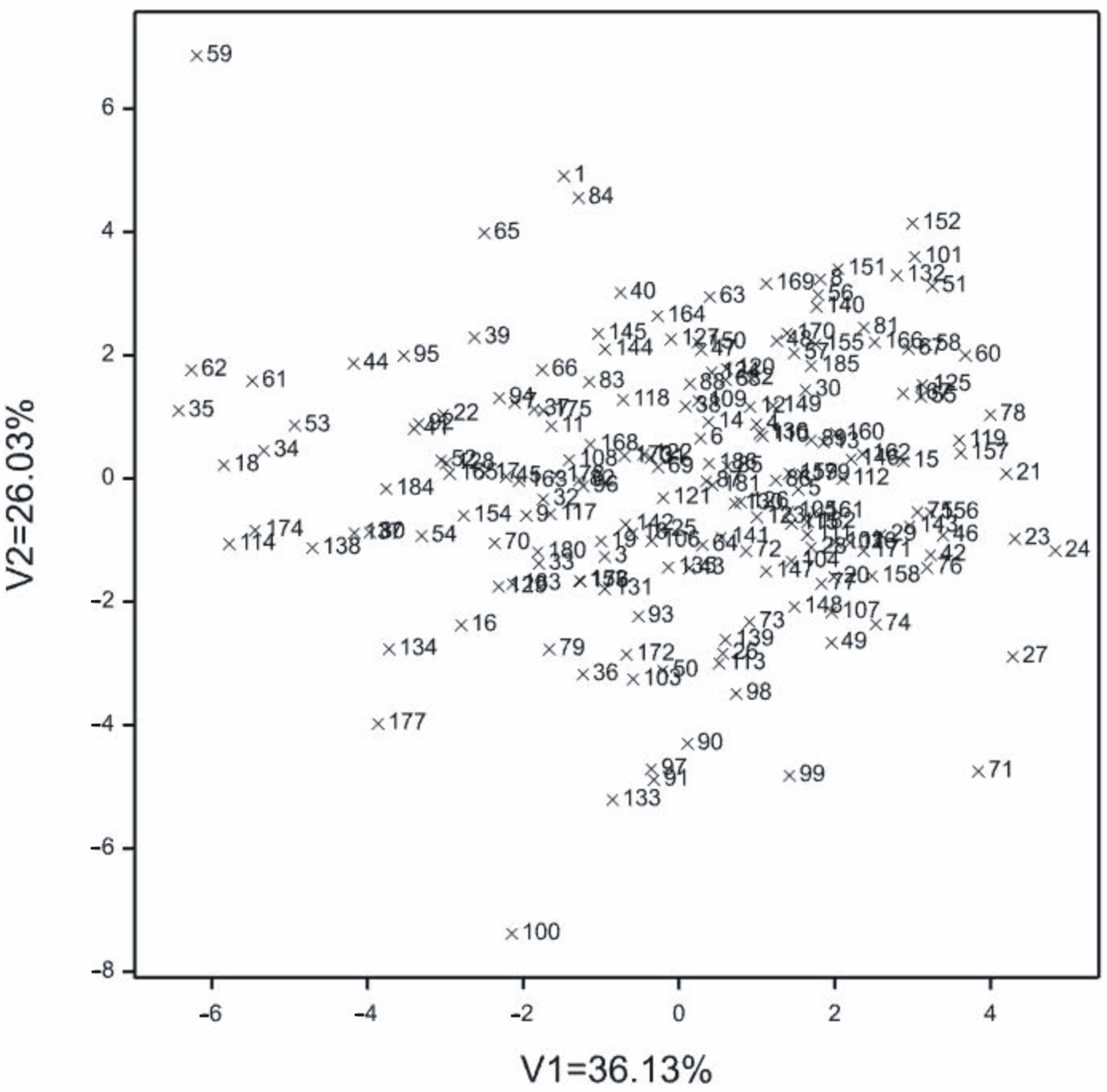
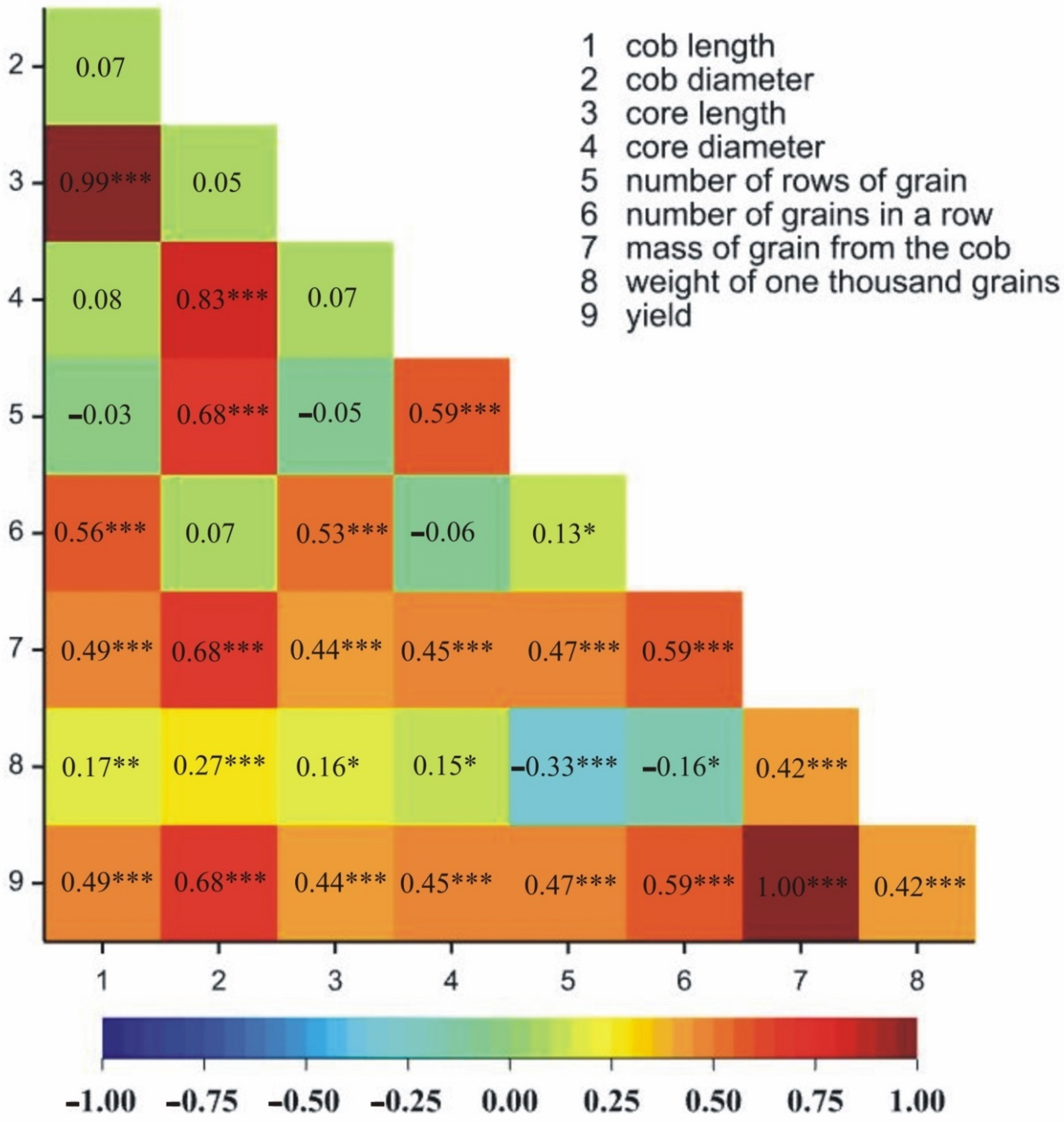
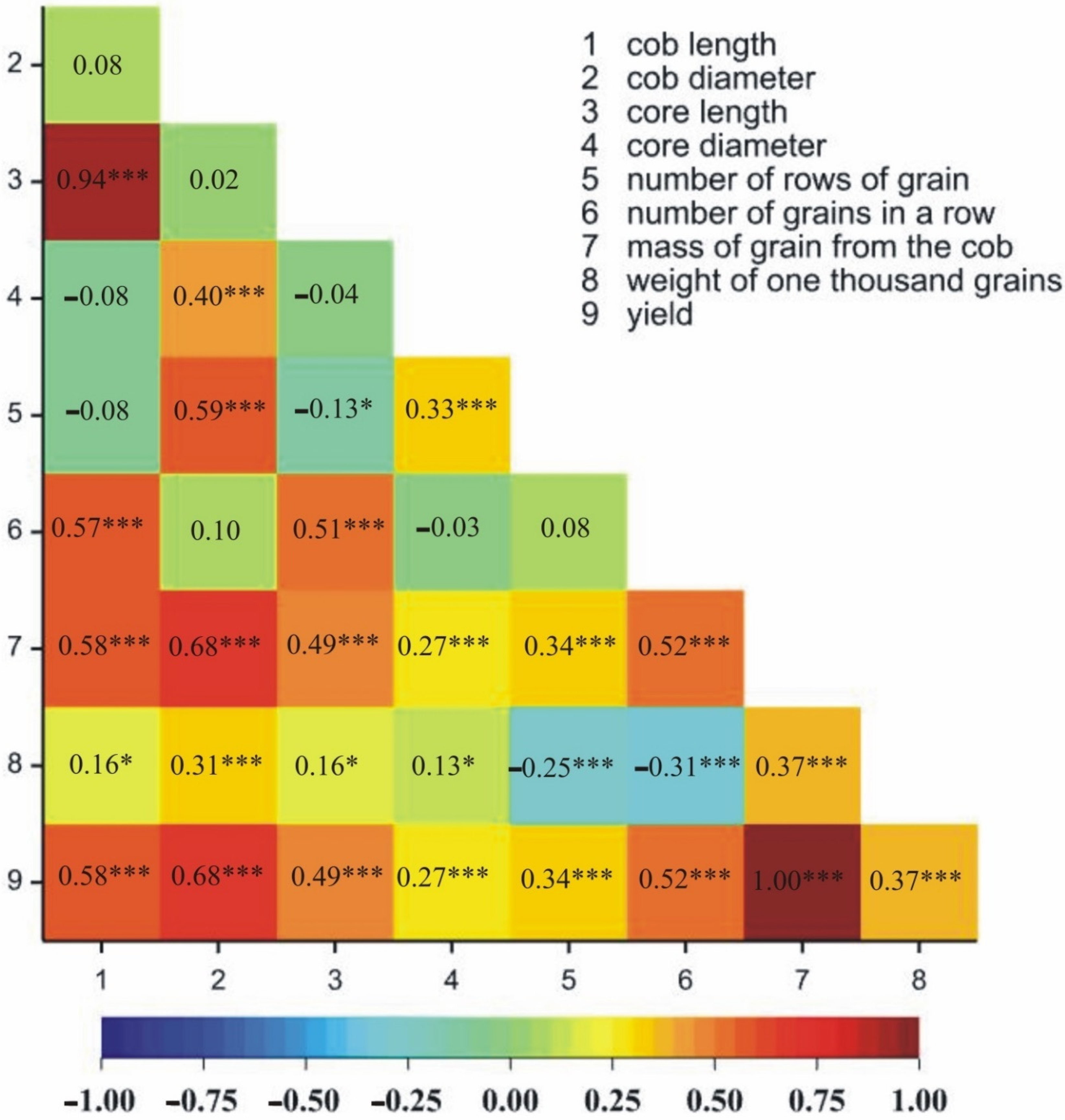
2.1.2. Relationships between Traits
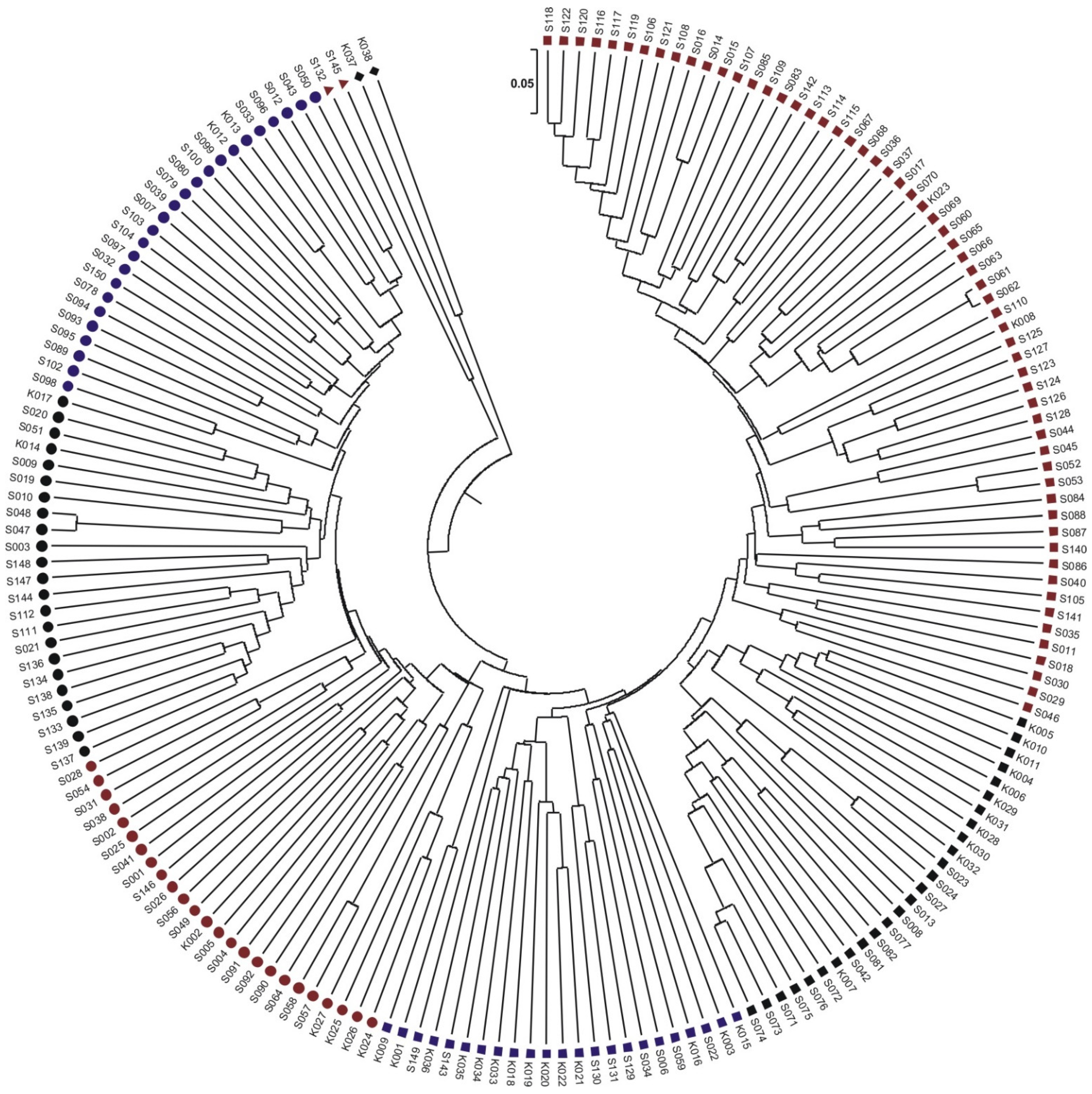
2.2. Next-Generation Sequencing to Identify SNP and SilicoDArT Markers Related to Maize Yield Genes
2.3. Association Mapping Using GWAS Analysis
2.4. Physical Mapping and Functional Analysis of Gene Sequences
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. Methods
4.2.1. Phenotyping
4.2.2. DNA Isolation
4.2.3. Genotyping
4.2.4. Statistical Analysis and Association Mapping Using GWAS Analysis
4.2.5. Association Mapping
4.2.6. Functional Analysis of Gene Sequences
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alexander, P.; Brown, C.; Arneth, A.; Finnigan, J.; Moran, D.; Rounsevella, M.D.A. Losses, inefficiencies and waste in the global food system. Agric. Syst. 2017, 153, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Opejin, A.K.; Aggarwal, R.M.; White, D.D.; Jones, J.L.; Maciejewski, R.; Mascaro, G.; Sarjoughian, H.S. A Bibliometric Analysis of Food-Energy-Water Nexus Literature. Sustainability 2020, 12, 1112. [Google Scholar] [CrossRef] [Green Version]
- Bekele, S.; Boddupalli, M.P.; Hellin, J.; Bänziger, M. Crops that feed the world 6. Past successes and future challenges to the role played by maize in global food security. Food Secur. 2011, 3, 307–327. [Google Scholar] [CrossRef] [Green Version]
- Bernal, J.S.; Dávila-Flores, A.M.; Medina, R.F.; Chen, Y.H.; Harrison, K.E.; Berrier, K.A. Did maize domestication and early spread mediate the population genetics of corn leafhopper? Insect Sci. 2019, 26, 569–586. [Google Scholar] [CrossRef] [Green Version]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, C.N.; Hirsch, C.D.; Brohammer, A.B.; Bowman, M.J.; Soifer, I.; Barad, O.; Shem-Tov, D.; Baruch, K.; Lu, F.; Hernandez, A.G.; et al. Draft, Assembly of Elite Inbred Line PH207 Provides Insights into Genomic and Transcriptome Diversity in Maize. Plant Cell 2016, 28, 2700–2714. [Google Scholar] [CrossRef]
- Springer, N.M.; Anderson, S.N.; Andorf, C.M.; Ahern, K.R.; Bai, F.; Barad, O.; Barbazuk, W.B.; Bass, H.W.; Baruch, K.; Ben-Zvi, G.; et al. The maize W22 genome provides a foundation for functional genomics and transposon biology. Nat. Genet. 2018, 50, 1282–1288. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Zhou, Y.; Chen, J.; Shi, J.; Zhao, H.; Zhao, H.; Song, W.; Zhang, M.; Cui, Y.; Dong, X.; et al. Extensive intraspecific gene order and gene structural variations between Mo17 and other maize genomes. Nat. Genet. 2018, 50, 1289–1295. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Liu, H.; Wu, L.; Warburton, M.; Yan, J. Genome-wide Association Studies in Maize: Praise and Stargaze. Mol. Plant 2017, 10, 359–374. [Google Scholar] [CrossRef] [Green Version]
- Tilman, D.; Cassman, K.G.; Matson, P.A. Agricultural sustainability and intensive production practices. Nature 2002, 418, 671–678. [Google Scholar] [CrossRef]
- Chen, F.; Fang, Z.; Gao, Q.; Youliang, Y.; Jia, L.; Yuan, L.; Mi, G.; Zhang, F. Evaluation of the yield and nitrogen use efficiency of the dominant maize hybrids grown in north and Northeast China. Sci. China Life Sci. 2013, 56, 552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, Y.; Peluso, P.; Shi, J.; Liang, T.; Stitzer, M.C.; Wang, B.; Campbell, M.S.; Stein, J.C.; Wei, X.; Chin, C.S.; et al. Improved maize reference genome with single-molecule technologies. Nature 2017, 546, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Peiffer, J.A.; Romay, M.C.; Gore, M.A.; Flint-Garcia, S.A.; Zhang, Z.; Millard, M.J.; Gardner, C.A.C.; McMullen, M.D.; Holland, J.B.; Bradbury, P.J.; et al. The genetic architecture of maize height. Genetics 2014, 196, 1337–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, J.; Ali, F.; Chen, G.; Li, H.; Mahuku, G.; Yang, N.; Narro, L.; Magorokosho, C.; Makumbi, D.; Yan, J. Genome-wide association mapping reveals novel sources of resistance to northern corn leaf blight in maize. BMC Plant Biol. 2015, 15, 206. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.D.; Perkins, A.; Williams, W.P.; Warburton, M.L. Using genome-wide associations to identify metabolic pathways involved in maize aflatoxin accumulation resistance. BMC Genom. 2015, 16, 673. [Google Scholar] [CrossRef] [Green Version]
- Nannas, N.J.; Dawe, R.K. Genetic and genomic toolbox of Zea mays. Genetics 2015, 199, 655–669. [Google Scholar] [CrossRef] [Green Version]
- Ben Ali, S.E.; Schamann, A.; Dobrovolny, S.; Indra, A.; Agapito-Tenfen, S.Z.; Hochegger, R.; Haslberger, A.G.; Brandes, C. Genetic and epigenetic characterization of the cry1Ab coding region and its 3′ flanking genomic region in MON810 maize using next-generation sequencing. Eur. Food Res. Technol. 2018, 244, 1473–1485. [Google Scholar] [CrossRef]
- Zaidi, P.H.; Seetharam, K.; Krishna, G.; Krishnamurthy, L.; Gajanan, S.; Babu, R.; Zerka, M.; Vinayan, M.T.; Vivek, B.S. Genomic regions associated with root traits under drought stress in tropical maize (Zea mays L.). PLoS ONE 2016, 11, e0164340. [Google Scholar] [CrossRef]
- Li, X.; Zhou, Z.; Ding, J.; Wu, Y.; Zhou, B.; Wang, R.; Ma, J.; Wang, S.; Zhang, X.; Xia, Z.; et al. Combined linkage and association mapping reveals QTL and candidate genes for plant and ear height in maize. Front. Plant Sci. 2016, 7, 833. [Google Scholar] [CrossRef] [Green Version]
- Muraya, M.M.; Chu, J.; Zhao, Y.; Junker, A.; Klukas, C.; Reif, J.C.; Altmann, T. Genetic variation of growth dynamics in maize (Zea mays L.) revealed through automated non-invasive phenotyping. Plant J. 2017, 89, 366–380. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.; Loladze, A.; Yuan, Y.; Wu, Y.; Zhang, A.; Chen, J.; Huestis, G.; Cao, J.; Chaikam, V.; Olsen, M.; et al. Genome-wide analysis of tar spot complex resistance in maize using genotyping-by-sequencing SNPs and whole-genome prediction. Plant Genome 2017, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araus, J.L.; Cairns, J.E. Field high-throughput phenotyping: The new crop breeding frontier. Trends Plant Sci. 2014, 19, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Godfray, H.C.J. How can 9-10 billion people be fed sustainably and equitably by 2050? In Is the Planet Full? Goldin, I., Ed.; Oxford University Press: Oxford, UK, 2014; pp. 104–120. [Google Scholar]
- Dell’Acqua, M.; Gatti, D.M.; Pea, G.; Cattonaro, F.; Coppens, F.; Magris, G.; Hlaing, A.L.; Aung, H.H.; Nelissen, H.; Baute, J.; et al. Genetic properties of the MAGIC maize population: A new platform for high definition QTL mapping in Zea mays. Genome Biol. 2015, 16, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef]
- Michael, T.P.; VanBuren, R. Progress, challenges and the future of crop genomes. Curr. Opin. Plant Biol. 2015, 24, 71–81. [Google Scholar] [CrossRef]
- Mahfouz, M.M.; Cardi, T.; Stewart, C.N. Next-generation precision genome engineering and plant biotechnology. Plant Cell Rep. 2016, 35, 1397–1399. [Google Scholar] [CrossRef]
- Pérez-de-Castro, A.M.; Vilanova, S.; Cañizares, J.; Pascual, L.; Blanca, J.M.; Díez, M.J.; Prohens, J.; Picó, B. Application of Genomic Tools in Plant Breeding. Curr. Genom. 2012, 13, 179–195. [Google Scholar] [CrossRef] [Green Version]
- Egan, A.N.; Schlueter, J.; Spooner, D.M. Applications of next-generation sequencing in plant biology. Am. J. Bot. 2012, 99, 175–185. [Google Scholar] [CrossRef] [Green Version]
- Tamasloukht, B.; Wong Quai Lam, M.S.-J.; Martinez, Y.; Tozo, K.; Barbier, O.; Jourda, C.; Jauneau, A.; Borderies, G.; Balzergue, S.; Renou, J.P.; et al. Characterization of a cinnamoyl-CoA reductase 1 (CCR1) mutant in maize: Effects on lignification, fibre development, and global gene expression. J. Exp. Bot. 2011, 62, 3837–3848. [Google Scholar] [CrossRef]
- Lauvergeat, V.; Lacomme, C.; Lacombe, E.; Lasserre, E.; Roby, D.; Grima-Pettenati, J. Two cinnamoyl-CoA reductase (CCR) genes from Arabidopsis thaliana are differentially expressed during development and in response to infection with pathogenic bacteria. Phytochemistry 2001, 57, 1187–1195. [Google Scholar] [CrossRef]
- Philippe, R.; Oana, D.; Reka, N.; Felten, J.; Corratge-Faillie, C.; Novak, O.; Morree, K.; Lacombe, B.; Martinez, Y.; Pfrunder, S.; et al. Arabidopsis WAT1 is a vacuolar auxin transport facilitator required for auxin homoeostasis. Nat. Commun. 2013, 4, 2625. [Google Scholar] [CrossRef]
- Raabe, K.; Honys, D.; Michailidis, C. Plant Physiology and Biochemistry The role of eukaryotic initiation factor 3 in plant translation regulation. Plant Physiol. Biochem. 2019, 145, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, Y.; Deguchi, M.; Youda, M.; Nishiguchi, M.; Nyunoya, H. Molecules and The Tomato Mosaic Tobamovirus Movement Protein Interacts with a Putative Transcriptional Coactivator KELP. Mol. Cells 2001, 12, 57–66. [Google Scholar] [PubMed]
- Oddy, J.; Raffan, S.; Wilkinson, M.D.; Elmore, J.S.; Halford, N.G. Understanding the Relationships between Free Asparagine in Grain and Other Traits to Breed Low-Asparagine Wheat. Plants 2022, 11, 669. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Wu, D.; Dai, H.; Sun, R.; Liu, A. Characterization and functional divergence of genes encoding sucrose in oilseeds castor bean. Oil Crop Sci. 2022, 7, 31–39. [Google Scholar] [CrossRef]
- Dhliwayo, T.; Pixley, K.; Menkir, A.; Warburton, M. Combining ability, genetic distances, and heterosis among elite CIMMYT and IITA tropical maize inbred lines. Crop Sci. 2009, 49, 1201–1210. [Google Scholar] [CrossRef]
- Chander, S.; Guo, Y.Q.; Yang, X.H.; Zhang, Z.; Lu, X.Q.; Yan, J.B.; Song, T.M.; Rocheford, T.R.; Li, J.S. Using molecular markers to identify two major loci controlling carotenoid contents in maize grain. Theor. Appl. Genet. 2008, 116, 223–233. [Google Scholar] [CrossRef]
- Choukan, R.; Hossainzadeh, A.; Ghannadha, M.R.; Warburton, M.L.; Talei, A.R.; Mohammadi, S.A. Use of SSR data to determine relationships and potential heterotic groupings within medium to late maturing Iranian maize inbred lines. Field Crops Res. 2006, 95, 212–222. [Google Scholar] [CrossRef]
- Babu, R.; Nair, S.K.; Kumar, A.; Rao, H.S.; Verma, P.; Gahalain, A.; Singh, I.S.; Gupta, H.S. Mapping QTLs for popping ability in a popcorn × flint corn cross. Theor. Appl. Genet. 2006, 112, 1392–1399. [Google Scholar] [CrossRef]
- Bernardo, R.; Yu, J. Prospects for genome-wide selection for quantitative traits in maize. Crop Sci. 2007, 47, 1082–1090. [Google Scholar] [CrossRef] [Green Version]
- Cuong, B.M.; Hao, P.X.; Regalado, E.; Van Truong, N. Genetic diversity of maize inbred lines revealed by SSR markers and their relationship with performance of F1 hybrids. In Proceedings of 9th Asian Regional Maize Workshop; Pixley, K., Zhang, S.H., Eds.; China Agricultural Science and Technology Press: Beijing, China, 2007; pp. 53–57. [Google Scholar]
- Tomkowiak, A.; Bocianowski, J.; Wolko, Ł.; Adamczyk, J.; Mikołajczyk, S.; Kowalczewski, Ł.P. Identification of Markers Associated with Yield Traits and Morphological Features in Maize (Zea mays L.). Plants 2019, 8, 330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilian, B.; Graner, A. NGS technologies for analyzing germplasm diversity in genebanks. Brief. Funct. Genom. 2012, 11, 38–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sansaloni, C.; Petroli, C.; Jaccoud, D.; Carling, J.; Detering, F.; Grattapaglia, D.; Kilian, A. Diversity Arrays Technology (DArT) and next-generation sequencing combined: Genome-wide, high throughput, highly informative genotyping for molecular breeding of Eucalyptus. BMC Proc. 2011, 5, 54. [Google Scholar] [CrossRef] [Green Version]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E.; Robust, A. Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nei, M.; Li, W.H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA 1979, 76, 5269–5273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starzycka-Korbas, E.; Weber, Z.; Matuszczak, M.; Bocianowski, J.; Budzianowski, G.; Stefanowicz, M.; Starzycki, M. The diversity of Sclerotinia sclerotiorum (Lib.) de Bary isolates from western Poland. J. Plant Pathol. 2021, 103, 185–195. [Google Scholar] [CrossRef]
- Seidler-Łożykowska, K.; Bocianowski, J. Evaluation of variability of morphological traits of selected caraway (Carum carvi L.) genotypes. Ind. Crops Prod. 2012, 35, 140–145. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Smolice | Kobierzyce | ||
|---|---|---|---|---|
| Trait | Mean | Standard Deviation | Mean | Standard Deviation |
| cob length (cm) | 13.1 | 1.5 | 15.4 | 1.7 |
| cob diameter (cm) | 4.0 | 0.3 | 3.9 | 0.4 |
| core length (cm) | 13.4 | 1.6 | 15.3 | 1.8 |
| core diameter (cm) | 2.2 | 0.4 | 2.1 | 0.3 |
| the number of rows of grain | 14.9 | 1.9 | 15.5 | 2.1 |
| the number of grains in a row | 25.0 | 4.5 | 27.6 | 3.4 |
| mass of grain from the cob (g) | 91.2 | 19.1 | 111.0 | 20.8 |
| weight of one thousand grains (g) | 284.0 | 46.7 | 260.0 | 41.5 |
| yield (kg) | 3.6 | 0.8 | 4.4 | 0.8 |
| Lines Number | Minimal Mahalanobis Distances | Lines Number | Maximal Mahalanobis Distances | ||
|---|---|---|---|---|---|
| 31 | 122 | 0.520 | 59 | 71 | 15.366 |
| 89 | 160 | 0.712 | 59 | 100 | 14.982 |
| 4 | 110 | 0.808 | 27 | 59 | 14.474 |
| 109 | 124 | 0.889 | 59 | 99 | 13.960 |
| 108 | 178 | 0.981 | 24 | 59 | 13.884 |
| 55 | 167 | 1.099 | 59 | 133 | 13.560 |
| 38 | 109 | 1.103 | 59 | 91 | 13.441 |
| 5 | 179 | 1.113 | 59 | 74 | 13.263 |
| 5 | 161 | 1.145 | 23 | 59 | 13.243 |
| 101 | 132 | 1.186 | 21 | 59 | 13.183 |
| 69 | 96 | 1.213 | 59 | 76 | 13.163 |
| 104 | 148 | 1.227 | 59 | 90 | 13.115 |
| 38 | 124 | 1.245 | 59 | 97 | 13.062 |
| 161 | 179 | 1.250 | 100 | 152 | 12.965 |
| 96 | 163 | 1.284 | 1 | 100 | 12.741 |
| 158 | 171 | 1.291 | 59 | 73 | 12.718 |
| 159 | 162 | 1.299 | 42 | 59 | 12.656 |
| Location | Trait | The Number of Significant Markers | Minimal Effect | Maximum Effect | Mean Effect | Total Effect | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DArT | SNP | Total | DArT | SNP | Total | DArT | SNP | Total | DArT | SNP | Total | DArT | SNP | Total | ||
| Kobierzyce | cob length | 136 | 1067 | 1203 | −1.234 | −1.469 | −1.469 | 1.381 | 1.574 | 1.574 | 0.092 | −0.043 | −0.028 | 12.483 | −46.064 | −33.581 |
| cob diameter | 185 | 1574 | 1759 | −0.279 | −0.305 | −0.305 | 0.269 | 0.311 | 0.311 | 0.039 | 0.008 | 0.011 | 7.146 | 12.990 | 20.136 | |
| core length | 132 | 1069 | 1201 | −1.270 | −1.497 | −1.497 | 1.404 | 1.586 | 1.586 | 0.117 | −0.026 | −0.010 | 15.389 | −27.567 | −12.178 | |
| core diameter | 236 | 2090 | 2326 | −0.215 | −0.227 | −0.227 | 0.225 | 0.247 | 0.247 | 0.043 | 0.027 | 0.029 | 10.225 | 57.202 | 67.427 | |
| the number of rows of grain | 29 | 292 | 321 | −1.194 | −1.358 | −1.358 | 1.256 | 1.526 | 1.526 | 0.047 | −0.046 | −0.038 | 1.374 | −13.482 | −12.108 | |
| the number of grains in a row | 8 | 122 | 130 | −1.727 | −2.145 | −2.145 | 1.727 | 2.276 | 2.276 | −0.413 | 0.029 | 0.001 | −3.301 | 3.489 | 0.188 | |
| mass of grain from the cob | 36 | 425 | 461 | −14.200 | −14.030 | −14.200 | 13.270 | 15.910 | 15.910 | 4.739 | 0.608 | 0.931 | 170.590 | 258.440 | 429.030 | |
| weight of one thousand grains | 56 | 462 | 518 | −28.630 | −30.290 | −30.290 | 29.920 | 27.620 | 29.920 | 2.466 | 0.904 | 1.073 | 138.110 | 417.540 | 555.650 | |
| yield | 37 | 424 | 461 | −0.568 | −0.561 | −0.568 | 0.531 | 0.637 | 0.637 | 0.195 | 0.023 | 0.037 | 7.227 | 9.941 | 17.168 | |
| Smolice | cob length | 140 | 1158 | 1298 | −1.020 | −1.265 | −1.265 | 1.254 | 1.297 | 1.297 | 0.050 | −0.056 | −0.044 | 6.970 | −64.653 | −57.683 |
| cob diameter | 147 | 1411 | 1558 | −0.240 | −0.263 | −0.263 | 0.213 | 0.237 | 0.237 | 0.008 | −0.004 | −0.003 | 1.131 | −5.495 | −4.365 | |
| core length | 137 | 1296 | 1433 | −1.051 | −1.290 | −1.290 | 1.309 | 1.354 | 1.354 | 0.003 | −0.094 | −0.085 | 0.423 | −121.678 | −121.255 | |
| core diameter | 24 | 246 | 270 | −0.211 | −0.234 | −0.234 | 0.200 | 0.241 | 0.241 | −0.017 | −0.015 | −0.015 | −0.413 | −3.675 | −4.088 | |
| the number of rows of grain | 54 | 428 | 482 | −1.221 | −1.455 | −1.455 | 1.105 | 1.439 | 1.439 | 0.293 | 0.126 | 0.144 | 15.840 | 53.761 | 69.601 | |
| the number of grains in a row | 42 | 435 | 477 | −2.755 | −3.273 | −3.273 | 2.523 | 2.662 | 2.662 | −0.520 | −1.048 | −1.001 | −21.844 | −455.778 | −477.622 | |
| mass of grain from the cob | 59 | 516 | 575 | −13.690 | −14.550 | −14.550 | 12.970 | 13.830 | 13.830 | 1.344 | −2.703 | −2.288 | 79.270 | −1394.760 | −1315.490 | |
| weight of one thousand grains | 42 | 319 | 361 | −26.780 | −30.830 | −30.830 | 28.510 | 33.290 | 33.290 | 0.946 | −1.444 | −1.166 | 39.730 | −460.480 | −420.750 | |
| yield | 59 | 516 | 575 | −0.548 | −0.582 | −0.582 | 0.519 | 0.553 | 0.553 | 0.054 | −0.108 | −0.092 | 3.170 | −55.796 | −52.626 | |
| Marker | Marker Type | Chromosome | Marker Location | Associated with | Candidate Genes |
|---|---|---|---|---|---|
| 17,300 | DArT | Chr1 | 2.15 × 108 | cob diameter, the number of rows of grain, mass of grain from the cob, yield | 40,523 bp at 5′ side: gdu1 |
| 68,570 bp at 3′ side: receptor-like protein kinase isoform | |||||
| 18,852 | DArT | Chr3 | 1.02 × 108 | cob diameter, the number of rows of grain, mass of grain from the cob, yield | 25,4293 bp at 5′ side: low quality protein: peptidyl-prolyl cis-trans isomerase |
| 18,563 bp at 3′ side: uncharacterized protein loc103650335 | |||||
| 1818 | DArT | Chr8 | 1.5 × 108 | cob diameter, the number of rows of grain, mass of grain from the cob, yield | A marker that is anchored to the gene cinnamoyl-CoA reductase 1 |
| 16,474 | DArT | Chr3 | 19,789,904 | cob length, cob diameter, core length, core diameter | 1270 bp at 5′ side: uncharacterized protein loc100382383 precursor |
| 4772 bp at 3′ side: uncharacterized protein loc100279241 precursor | |||||
| 14,506 | DArT | Chr9 | 28,978,769 | cob diameter, the number of rows of grain, mass of grain from the cob, yield | A marker that is anchored (WAT1-related protein At1g09380) |
| 13,517 | DArT | Chr9 | 1.31 × 108 | cob length, core length, the number of rows of grain, weight of one thousand grains | 8016 bp at 5′ side: uncharacterized protein loc103639077 isoform 1 |
| 450 bp at 3′ side: allene-oxide cyclase2 | |||||
| 2317 | DArT | Chr7 | 1.38 × 108 | cob diameter, the number of rows of grain, mass of grain from the cob, yield | A marker that is anchored (eukaryotic translation initiation factor 3 subunit c) |
| 7950 | DArT | Chr2 | 43,524,954 | cob diameter, the number of rows of grain, mass of grain from the cob, yield | 233,907 bp at 5′ side: actin binding protein precursor |
| 5461 bp at 3′ side: mads-box transcription factor 27 isoform 2 | |||||
| 16,703 | DArT | Chr2 | 1.68 × 108 | cob diameter, the number of rows of grain, mass of grain from the cob, yield | A marker that is anchored (uncharacterized protein loc100282883 |
| 17,490 | DArT | Chr10 | 1.39 × 108 | cob length, cob diameter, core length, the number of rows of grain | 91,320 bp at 5′ side: scarecrow-like protein 8 |
| 6776 bp at 3′ side: uncharacterized protein loc100383502 | |||||
| 17,843 | DArT | Chr3 | 2.25 × 108 | cob length, the number of grains in a row, mass of grain from the cob, yield | 1290 bp at 5′ side: uncharacterized protein loc100192921 isoform 1 |
| 6791 bp at 3′ side: uncharacterized protein loc100276743 | |||||
| 18,664 | DArT | Chr5 | 2.11 × 108 | cob diameter, the number of rows of grain, mass of grain from the cob, yield | 85,540 bp at 5′ side: uncharacterized protein loc100278506 |
| 101,692 bp at 3′ side: delta-12 fatty acid desaturasefad2 isoform 1 | |||||
| 3233 | DArT | Chr3 | 2.1 × 108 | cob diameter, the number of rows of grain, mass of grain from the cob, yield | A marker that is anchored RNA polymerase II transcriptional coactivator KELP |
| 4205 | DArT | Chr5 | 2.26 × 108 | cob diameter, the number of rows of grain, mass of grain from the cob, yield | 42,608 bp at 5′ side: uncharacterized protein loc118472127 |
| 46,806 bp at 3′ side: callose synthase | |||||
| 11,657 | DArT | Chr5 | 2.22 × 108 | cob diameter, core diameter, mass of grain from the cob, yield | A marker that is anchored aspartate aminotransferase |
| 12,812 | DArT | Chr1 | 15,198,950 | cob length, core length, the number of rows of grain, weight of one thousand grains | A marker that is anchored sucrose transporter 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomkowiak, A.; Nowak, B.; Sobiech, A.; Bocianowski, J.; Wolko, Ł.; Spychała, J. The Use of DArTseq Technology to Identify New SNP and SilicoDArT Markers Related to the Yield-Related Traits Components in Maize. Genes 2022, 13, 848. https://doi.org/10.3390/genes13050848
Tomkowiak A, Nowak B, Sobiech A, Bocianowski J, Wolko Ł, Spychała J. The Use of DArTseq Technology to Identify New SNP and SilicoDArT Markers Related to the Yield-Related Traits Components in Maize. Genes. 2022; 13(5):848. https://doi.org/10.3390/genes13050848
Chicago/Turabian StyleTomkowiak, Agnieszka, Bartosz Nowak, Aleksandra Sobiech, Jan Bocianowski, Łukasz Wolko, and Julia Spychała. 2022. "The Use of DArTseq Technology to Identify New SNP and SilicoDArT Markers Related to the Yield-Related Traits Components in Maize" Genes 13, no. 5: 848. https://doi.org/10.3390/genes13050848
APA StyleTomkowiak, A., Nowak, B., Sobiech, A., Bocianowski, J., Wolko, Ł., & Spychała, J. (2022). The Use of DArTseq Technology to Identify New SNP and SilicoDArT Markers Related to the Yield-Related Traits Components in Maize. Genes, 13(5), 848. https://doi.org/10.3390/genes13050848