
A Novel Strategy to Identify Prognosis-Relevant Gene Sets in Cancers



Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Acquisition
2.2. Using GSEA to Detect Enrichment of Single Gene Prognostic Signals
2.3. Gene Set Survival Analysis
2.4. Meta Differential Coexpression Analysis
2.5. Gene Set Crosstalk
3. Results
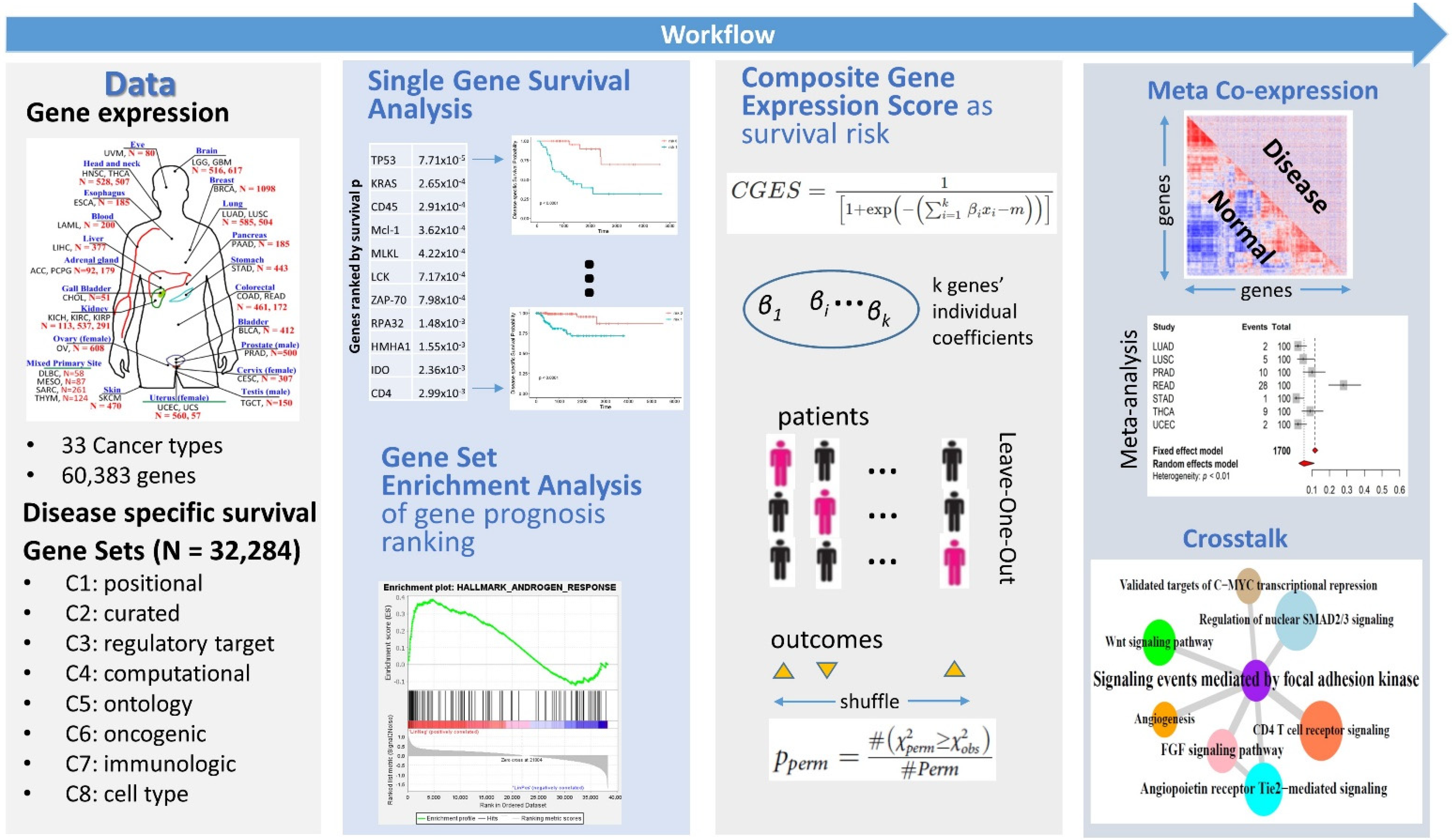
3.1. Overall Strategy
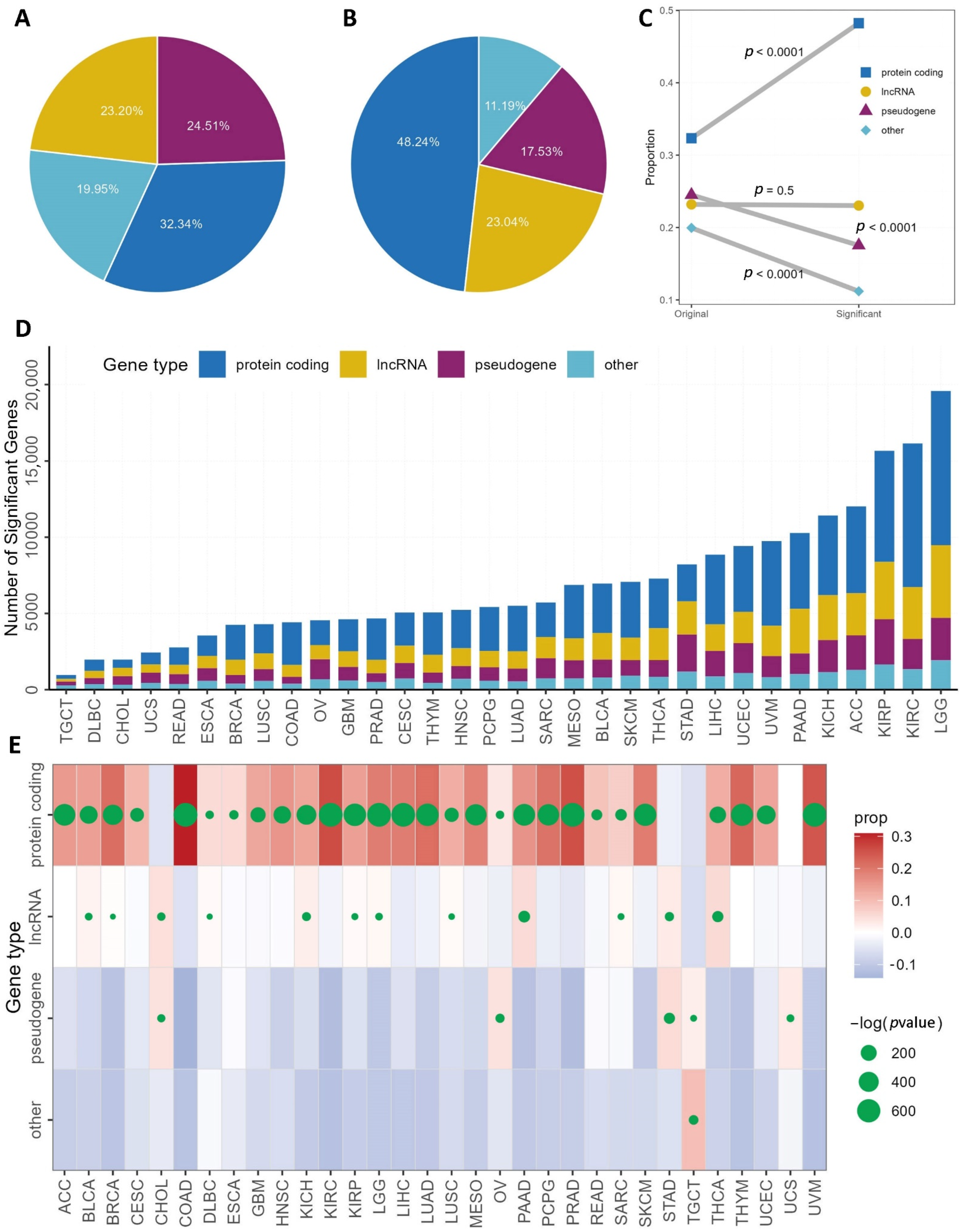
3.2. Single Gene Survival
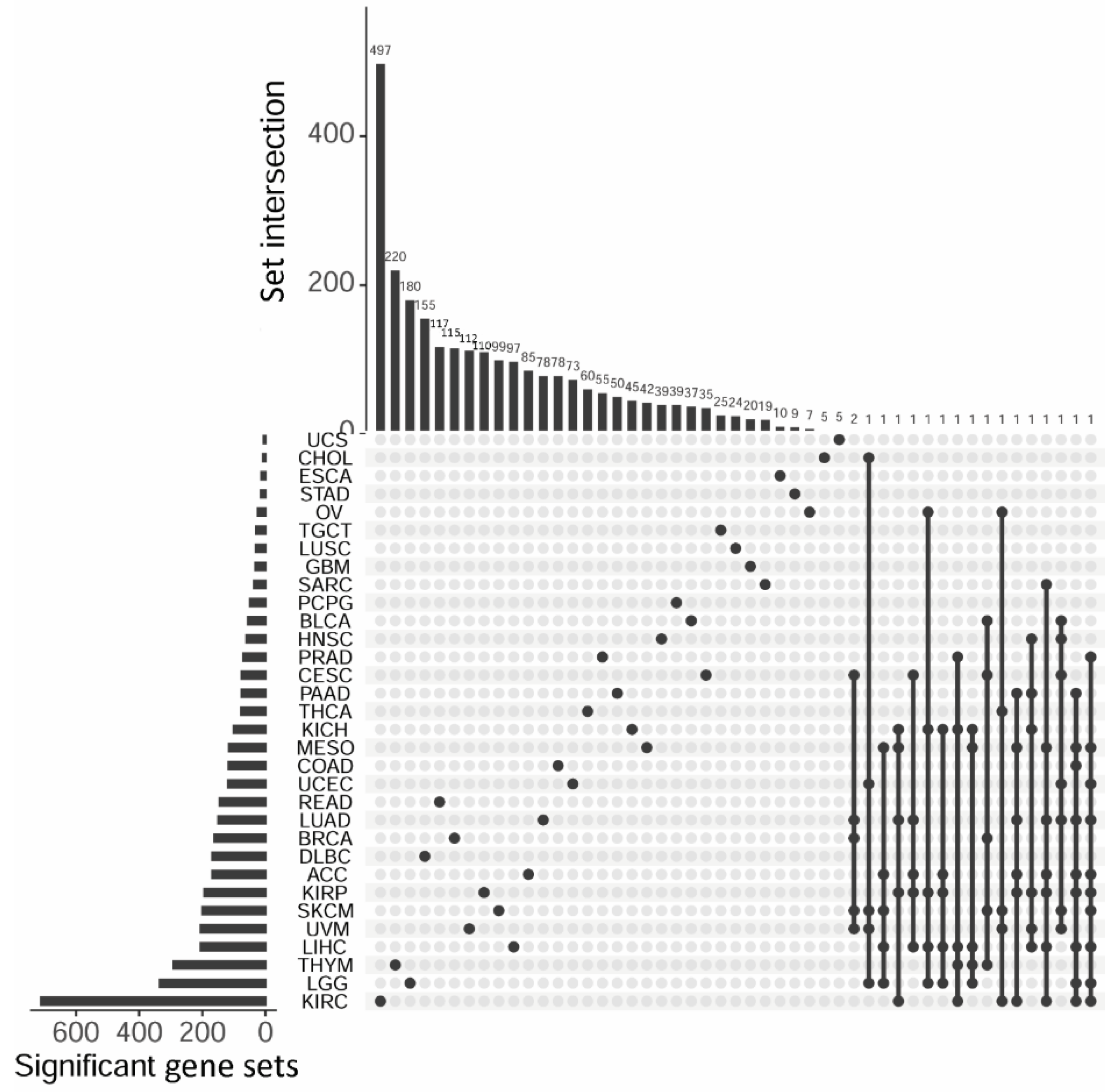
3.3. Gene Set Survival Analysis
3.4. Pan-Cancer Differential Coexpression of Gene Sets
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hui, D.; Mo, L.; Paiva, C.E. The Importance of Prognostication: Impact of Prognostic Predictions, Disclosures, Awareness, and Acceptance on Patient Outcomes. Curr. Treat. Options Oncol. 2021, 22, 12. [Google Scholar] [CrossRef] [PubMed]
- Freedman, R.A.; Keating, N.L.; Lin, N.U.; Winer, E.P.; Vaz-Luis, I.; Lii, J.; Exman, P.; Barry, W.T. Breast cancer-specific survival by age: Worse outcomes for the oldest patients. Cancer-Am. Cancer Soc. 2018, 124, 2184–2191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, L.; Canchola, A.J.; Spiegel, D.; Ladabaum, U.; Haile, R.; Gomez, S.L. Racial and Ethnic Disparities in Cancer Survival: The Contribution of Tumor, Sociodemographic, Institutional, and Neighborhood Characteristics. J. Clin. Oncol. 2018, 36, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Ping, J.; Oyebamiji, O.; Yu, H.; Ness, S.; Chien, J.; Ye, F.; Kang, H.; Samuels, D.; Ivanov, S.; Chen, D.; et al. MutEx: A multifaceted gateway for exploring integrative pan-cancer genomic data. Brief Bioinform. 2020, 21, 1479–1486. [Google Scholar] [CrossRef] [PubMed]
- Ballman, K.V. Biomarker: Predictive or Prognostic? J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 3968–3971. [Google Scholar] [CrossRef]
- Louie, A.D.; Huntington, K.; Carlsen, L.; Zhou, L.; El-Deiry, W.S. Integrating Molecular Biomarker Inputs Into Development and Use of Clinical Cancer Therapeutics. Front. Pharmacol. 2021, 12, 747194. [Google Scholar] [CrossRef]
- Slodkowska, E.A.; Ross, J.S. MammaPrint 70-gene signature: Another milestone in personalized medical care for breast cancer patients. Expert Rev. Mol. Diagn. 2009, 9, 417–422. [Google Scholar] [CrossRef]
- Paik, S.; Shak, S.; Tang, G.; Kim, C.; Baker, J.; Cronin, M.; Baehner, F.L.; Walker, M.G.; Watson, D.; Park, T.; et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N. Engl. J. Med. 2004, 351, 2817–2826. [Google Scholar] [CrossRef] [Green Version]
- Molina-Pinelo, S.; Pastor, M.D.; Paz-Ares, L. VeriStrat: A prognostic and/or predictive biomarker for advanced lung cancer patients? Expert Rev. Respir. Med. 2014, 8, 1–4. [Google Scholar] [CrossRef]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef]
- Ooi, H.S.; Schneider, G.; Lim, T.; Chan, Y.; Eisenhaber, B.; Eisenhaber, F. Biomolecular Pathway Databases. In Data Mining Techniques for the Life Sciences; Carugo, O., Eisenhaber, F., Eds.; Humana Press: Totowa, NJ, USA, 2010. [Google Scholar] [CrossRef]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdottir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, B.; Shi, J.; Kang, H.; Oyebamiji, O.; Hill, D.; Yu, H.; Ness, S.; Ye, F.; Ping, J.; He, J.; et al. Advancing Pan-cancer Gene Expression Survial Analysis by Inclusion of Non-coding RNA. RNA Biol. 2020, 17, 1666–1673. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korotkevich, G.; Sukhov, V.; Sergushichev, A. Fast gene set enrichment analysis. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- van Dam, S.; Vosa, U.; van der Graaf, A.; Franke, L.; de Magalhaes, J.P. Gene co-expression analysis for functional classification and gene-disease predictions. Brief. Bioinform. 2018, 19, 575–592. [Google Scholar] [CrossRef]
- Lea, A.; Subramaniam, M.; Ko, A.; Lehtimaki, T.; Raitoharju, E.; Kahonen, M.; Seppala, I.; Mononen, N.; Raitakari, O.T.; Ala-Korpela, M.; et al. Genetic and environmental perturbations lead to regulatory decoherence. eLife 2019, 8, e40538. [Google Scholar] [CrossRef]
- Guo, Y.; Yu, H.; Song, H.; He, J.; Oyebamiji, O.; Kang, H.; Ping, J.; Ness, S.; Shyr, Y.; Ye, F. MetaGSCA: A tool for meta-analysis of gene set differential coexpression. PLoS Comput. Biol. 2021, 17, e1008976. [Google Scholar] [CrossRef]
- Rahmatallah, Y.; Emmert-Streib, F.; Glazko, G. Gene Sets Net Correlations Analysis (GSNCA): A multivariate differential coexpression test for gene sets. Bioinformatics 2014, 30, 360–368. [Google Scholar] [CrossRef]
- Yang, J.; Yu, H.; Liu, B.H.; Zhao, Z.; Liu, L.; Ma, L.X.; Li, Y.X.; Li, Y.Y. DCGL v2.0: An R package for unveiling differential regulation from differential co-expression. PLoS ONE 2013, 8, e79729. [Google Scholar] [CrossRef]
- Yu, H.; Liu, B.H.; Ye, Z.Q.; Li, C.; Li, Y.X.; Li, Y.Y. Link-based quantitative methods to identify differentially coexpressed genes and gene pairs. BMC Bioinform. 2011, 12, 315. [Google Scholar] [CrossRef] [Green Version]
- Barabasi, A.L.; Oltvai, Z.N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004, 5, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Chen, D.; Oyebamiji, O.; Zhao, Y.Y.; Guo, Y. Expression correlation attenuates within and between key signaling pathways in chronic kidney disease. BMC Med. Genom. 2020, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Guo, Y.; Chen, J.; Chen, X.; Jia, P.; Zhao, Z. Rewired Pathways and Disrupted Pathway Crosstalk in Schizophrenia Transcriptomes by Multiple Differential Coexpression Methods. Genes 2021, 12, 665. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, S. Detection of characteristic sub pathway network for angiogenesis based on the comprehensive pathway network. BMC Bioinform. 2010, 11 (Suppl. 1), S32. [Google Scholar] [CrossRef] [Green Version]
- Montero-Conde, C.; Martin-Campos, J.M.; Lerma, E.; Gimenez, G.; Martinez-Guitarte, J.L.; Combalia, N.; Montaner, D.; Matias-Guiu, X.; Dopazo, J.; de Leiva, A.; et al. Molecular profiling related to poor prognosis in thyroid carcinoma. Combining gene expression data and biological information. Oncogene 2008, 27, 1554–1561. [Google Scholar] [CrossRef] [Green Version]
- Crosby, M.E.; Jacobberger, J.; Gupta, D.; Macklis, R.M.; Almasan, A. E2F4 regulates a stable G2 arrest response to genotoxic stress in prostate carcinoma. Oncogene 2007, 26, 1897–1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finetti, P.; Cervera, N.; Charafe-Jauffret, E.; Chabannon, C.; Charpin, C.; Chaffanet, M.; Jacquemier, J.; Viens, P.; Birnbaum, D.; Bertucci, F. Sixteen-kinase gene expression identifies luminal breast cancers with poor prognosis. Cancer Res. 2008, 68, 767–776. [Google Scholar] [CrossRef] [Green Version]
- Kumamoto, K.; Spillare, E.A.; Fujita, K.; Horikawa, I.; Yamashita, T.; Appella, E.; Nagashima, M.; Takenoshita, S.; Yokota, J.; Harris, C.C. Nutlin-3a activates p53 to both down-regulate inhibitor of growth 2 and up-regulate mir-34a, mir-34b, and mir-34c expression, and induce senescence. Cancer Res. 2008, 68, 3193–3203. [Google Scholar] [CrossRef] [Green Version]
- Anglani, R.; Creanza, T.M.; Liuzzi, V.C.; Piepoli, A.; Panza, A.; Andriulli, A.; Ancona, N. Loss of connectivity in cancer co-expression networks. PLoS ONE 2014, 9, e87075. [Google Scholar] [CrossRef]
- Dalgic, E.; Konu, O.; Oz, Z.S.; Chan, C. Lower connectivity of tumor coexpression networks is not specific to cancer. Silico Biol. 2019, 13, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charkhchi, P.; Cybulski, C.; Gronwald, J.; Wong, F.O.; Narod, S.A.; Akbari, M.R. CA125 and Ovarian Cancer: A Comprehensive Review. Cancers 2020, 12, 3730. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, T.; Bianchini, G.; Booser, D.; Qi, Y.; Coutant, C.; Shiang, C.Y.; Santarpia, L.; Matsuoka, J.; Hortobagyi, G.N.; Symmans, W.F.; et al. Gene pathways associated with prognosis and chemotherapy sensitivity in molecular subtypes of breast cancer. J. Natl. Cancer Inst. 2011, 103, 264–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efron, B.; Tibshirani, R. On testing the significance of sets of genes. Ann. Appl. Stat. 2007, 1, 107–129. [Google Scholar] [CrossRef] [Green Version]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C.; et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009, 462, 108–112. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Y.; Akinyemiju, T.; Ojesina, A.I.; Buckhaults, P.; Liu, N.; Xu, B.; Yi, N. Pathway-Structured Predictive Model for Cancer Survival Prediction: A Two-Stage Approach. Genetics 2017, 205, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Dereli, O.; Oguz, C.; Gonen, M. Path2Surv: Pathway/gene set-based survival analysis using multiple kernel learning. Bioinformatics 2019, 35, 5137–5145. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Gene Set Name | # Genes | p |
|---|---|---|---|
| C2 | MONTERO THYROID CANCER POOR SURVIVAL UP | 12 | 0.021 |
| C2 | FINETTI BREAST CANCER KINOME RED | 16 | 0.022 |
| C2 | KUMAMOTO RESPONSE TO NUTLIN 3A DN | 9 | 0.023 |
| C5 | GOBP DOUBLE STRAND BREAK REPAIR VIA BREAK INDUCED REPLICATION | 12 | 0.025 |
| C5 | GOCC CONDENSED CHROMOSOME INNER KINETOCHORE | 5 | 0.029 |
| C4 | MODULE 315 | 16 | 0.031 |
| C2 | CROSBY E2F4 TARGETS | 6 | 0.031 |
| C5 | GOCC MHC CLASS II PROTEIN COMPLEX | 14 | 0.060 |
| C5 | GOCC α DNA POLYMERASE PRIMASE COMPLEX | 5 | 0.061 |
| C2 | REACTOME G2 M DNA REPLICATION CHECKPOINT | 5 | 0.063 |
| C2 | BIOCARTA TCRA PATHWAY | 12 | 0.070 |
| C7 | MATSUMIYA PBMC MODIFIED VACCINIA ANKARA VACCINE AGE 4 6MO BCG PRIMED 28DY UP | 9 | 0.091 |
| C5 | GOCC NUCLEAR INCLUSION BODY | 11 | 0.117 |
| C5 | GOCC EUKARYOTIC TRANSLATION INITIATION FACTOR 2 COMPLEX | 4 | 0.132 |
| C5 | GOMF MHC CLASS II RECEPTOR ACTIVITY | 9 | 0.158 |
| C5 | GOCC ALPHA BETA T CELL RECEPTOR COMPLEX | 7 | 0.161 |
| C5 | GOMF D LOOP DNA BINDING | 5 | 0.182 |
| C7 | ANDERSON BLOOD CN54GP140 ADJUVANTED WITH GLA AF AGE 18 45YO HIGH IGM RESPONDERS 3DY 7D DN | 6 | 0.254 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pu, J.; Yu, H.; Guo, Y. A Novel Strategy to Identify Prognosis-Relevant Gene Sets in Cancers. Genes 2022, 13, 862. https://doi.org/10.3390/genes13050862
Pu J, Yu H, Guo Y. A Novel Strategy to Identify Prognosis-Relevant Gene Sets in Cancers. Genes. 2022; 13(5):862. https://doi.org/10.3390/genes13050862
Chicago/Turabian StylePu, Junyi, Hui Yu, and Yan Guo. 2022. "A Novel Strategy to Identify Prognosis-Relevant Gene Sets in Cancers" Genes 13, no. 5: 862. https://doi.org/10.3390/genes13050862
APA StylePu, J., Yu, H., & Guo, Y. (2022). A Novel Strategy to Identify Prognosis-Relevant Gene Sets in Cancers. Genes, 13(5), 862. https://doi.org/10.3390/genes13050862