
A Clinical Case of a Homozygous Deletion in the APOA5 Gene with Severe Hypertriglyceridemia

 , , ,
, , ,  , ,
, ,
Abstract
:1. Introduction
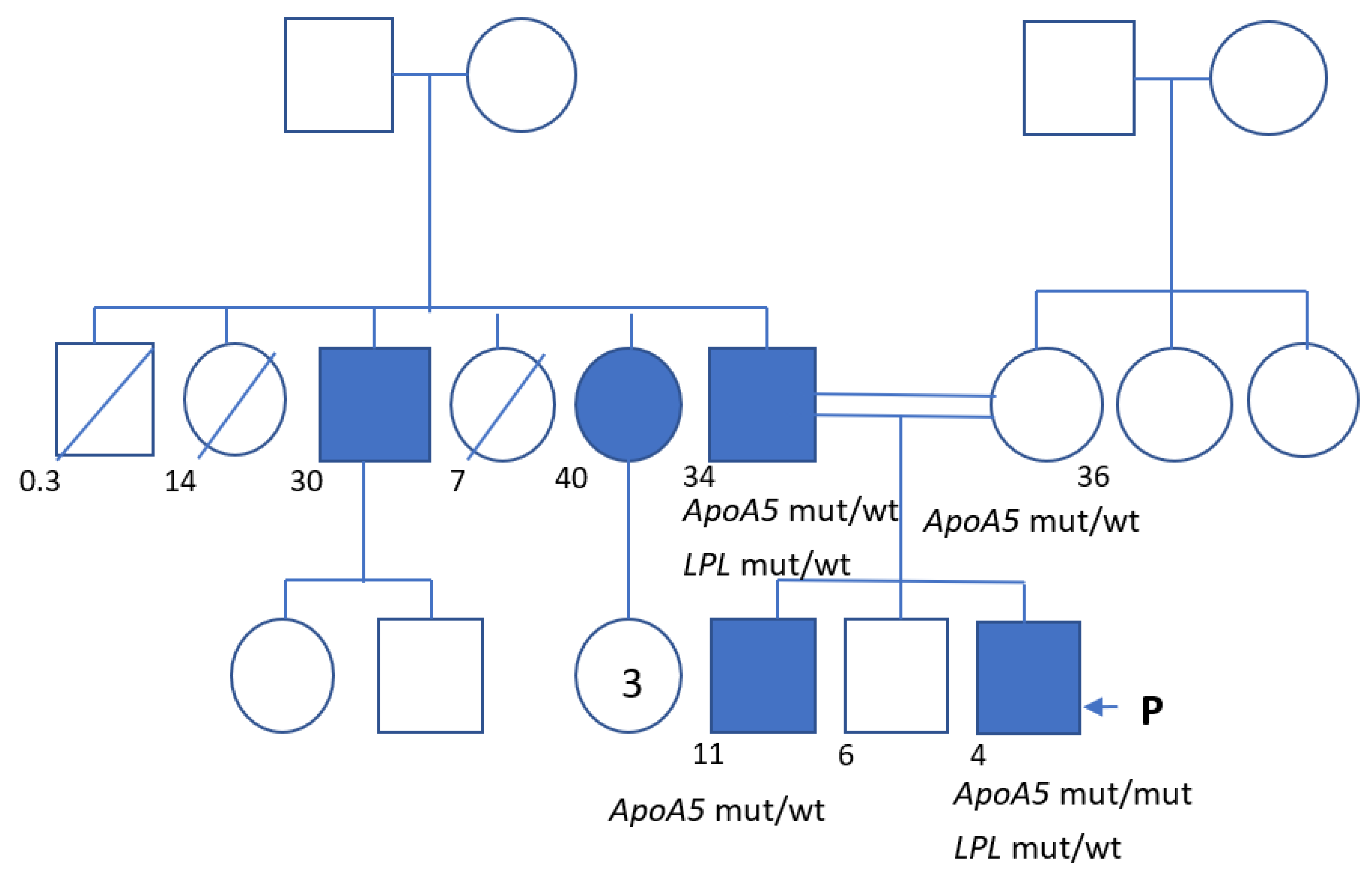
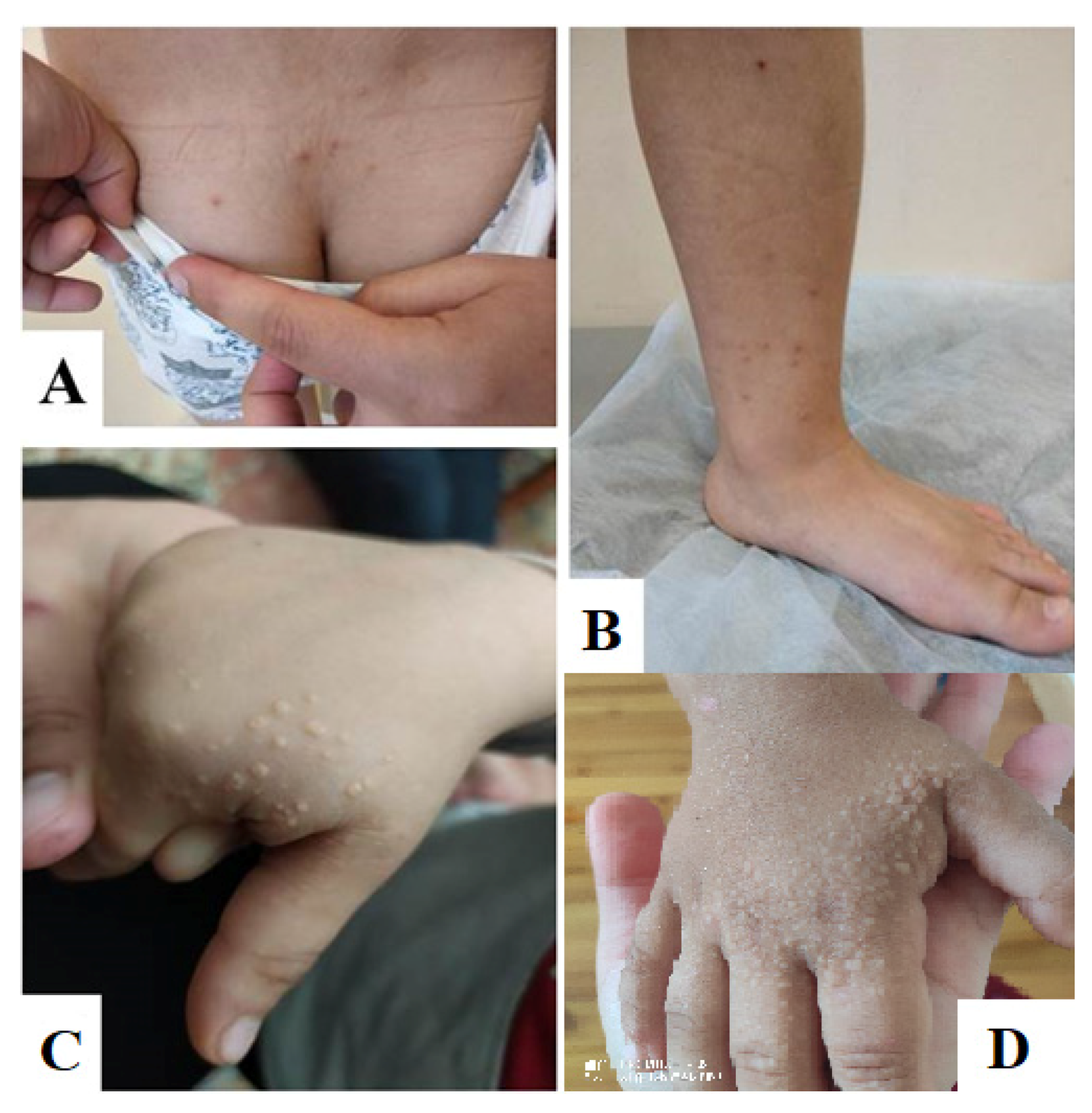
2. Clinical Case
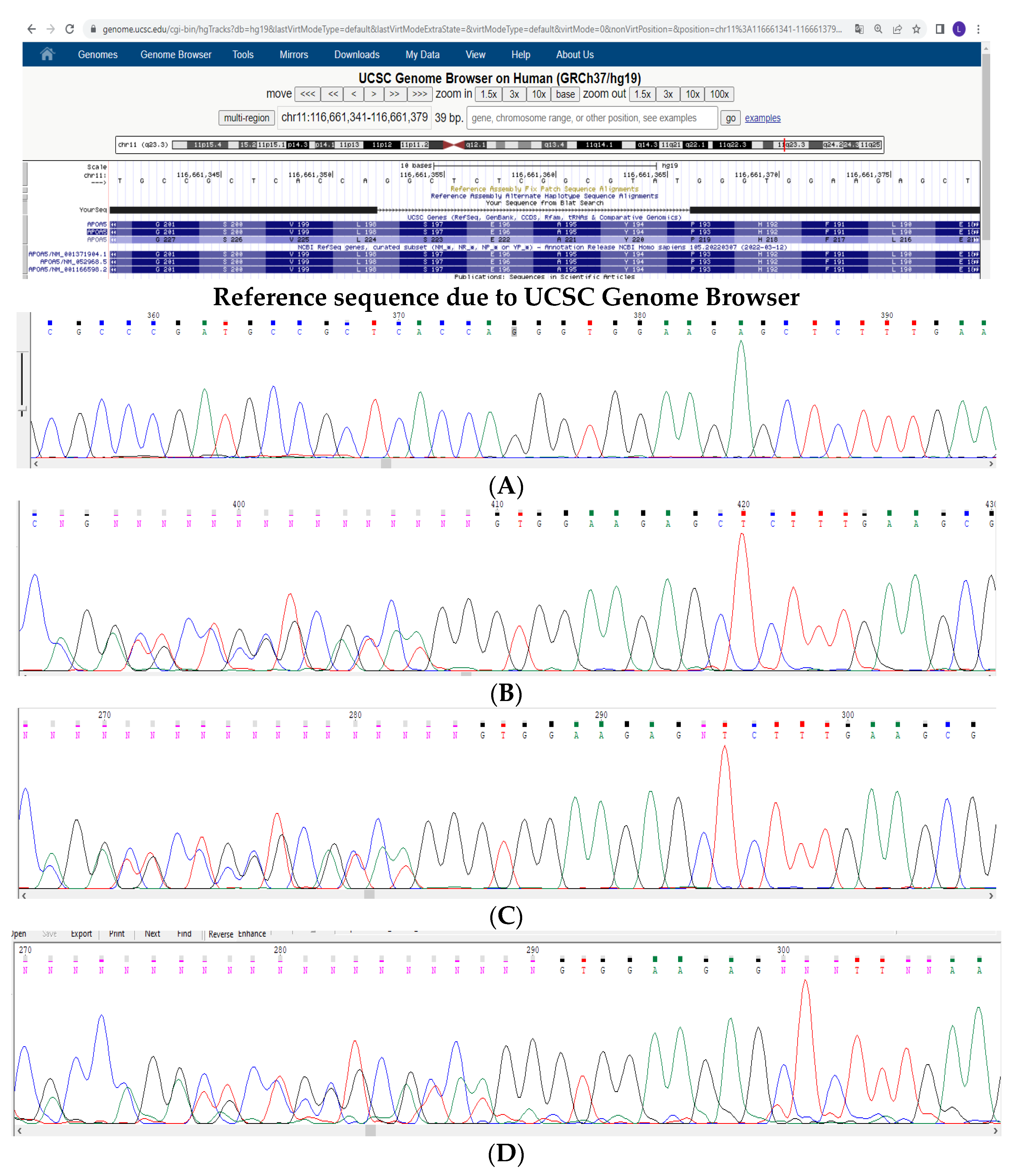
3. Materials and Methods
4. Results and Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Miller, M.; Stone, N.J.; Ballantyne, C.; Bittner, V.; Criqui, M.H.; Ginsberg, H.N.; Goldberg, A.C.; Howard, W.J.; Jacobson, M.S.; Kris-Etherton, P.M.; et al. Triglycerides and cardiovascular disease: A scientific statement from the American Heart Association. Circulation 2011, 123, 2292–2333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; De Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 AHA/ACC/AACVPR/ AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 139, e1082–e1143. [Google Scholar] [PubMed]
- Christian, J.B.; Bourgeois, N.; Snipes, R.; Lowe, K.A. Prevalence of severe (500 to 2,000 mg/dl) hypertriglyceridemia in United States adults. Am. J. Cardiol. 2011, 107, 891–897. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Tóth, P.P.; Potter, D.; Ming, E.E. Prevalence of lipid abnormalities in the United States: The National Health and Nutrition Examination Survey 2003–2006. J. Clin. Lipidol. 2012, 6, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.; Cantor, R.M.; Weissglas-Volkov, D.; Nikkola, E.; Reddy, P.M.; Sinsheimer, J.S.; Pasaniuc, B.; Brown, R.; Alvarez, M.; Rodriguez, A.; et al. Amerindian-specific regions under positive selection harbour new lipid variants in Latinos. Nat Commun. 2014, 5, 3983. [Google Scholar] [CrossRef] [Green Version]
- Meshkov, A.N.; Ershova, A.I.; Deev, A.D.; Metelskaya, V.A.; Zhernakova, Y.u.V.; Rotar, O.P.; Shalnova, S.A.; Boytsov, S.A. Distribution of Lipid Profile Values in Economically Active Men and Women in Russian Federation: Results of The Esse-Rf Study for the Years 2012–2014. Cardiovasc. Ther. Prev. 2017, 16, 62–67. [Google Scholar] [CrossRef]
- Castelli, W.P. Epidemiology of triglycerides: A view from Framingham. Am. J. Cardiol. 1992, 70, H3–H9. [Google Scholar] [CrossRef]
- Karlson, B.W.; Palmer, M.K.; Nicholls, S.J.; Lundman, P.; Barter, P.J. A VOYAGER meta-analysis of the impact of statin therapy on low-density lipoprotein cholesterol and triglyceride levels in patients with hypertriglyceridemia. Am. J. Cardiol. 2016, 117, 1444–1448. [Google Scholar] [CrossRef]
- Rhodes, K.S.; Weintraub, M.; Marchlewicz, E.H.; Rubenfire, M.; Brook, R.D. Medical nutrition therapy is the essential cornerstone for effective treatment of ‘refractory’ severe hypertriglyceridemia regardless of pharmaceutical treatment: Evidence from a lipid management program. J. Clin. Lipidol. 2015, 9, 559–567. [Google Scholar] [CrossRef]
- Freiberg, J.J.; Tybjaerg-Hansen, A.; Jensen, J.S.; Nordestgaard, B.G. Nonfasting triglycerides and risk of ischemic stroke in the general population. JAMA 2008, 300, 2142–2152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, V.J.; Bishop, L.; Laranjo, N.; Harshfield, B.J.; Kwiat, C.; Sacks, F.M. Contribution of High Plasma Triglycerides and Low High-Density Lipoprotein Cholesterol to Residual Risk of Coronary Heart Disease After Establishment of Low-Density Lipoprotein Cholesterol Control. Am. J. Cardiol. 2010, 106, 757–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reaven, G.M.; Chen, Y.D.; Jeppesen, J.; Maheux, P.; Krauss, R.M. Insulin resistance and hyperinsulinemia in individuals with small, dense low density lipoprotein particles. J. Clin. Investig. 1993, 92, 141–146. [Google Scholar] [CrossRef] [Green Version]
- Hegele, R.A.; Ginsberg, H.N.; Chapman, M.J.; Nordestgaard, B.G.; Kuivenhoven, J.A.; Averna, M.; Borén, J.; Bruckert, E.; Catapano, A.L.; Descamps, O.S.; et al. The polygenic nature of hypertriglyceridaemia: Implications for definition, diagnosis, and management. Lancet Diabetes Endocrinol. 2014, 2, 655–666. [Google Scholar] [CrossRef] [Green Version]
- Garg, R.; Rustagi, T. Management of Hypertriglyceridemia Induced Acute Pancreatitis. Biomed Res. Int. 2018, 2018, 4721357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldhaleei, W.A.; Alnuaimi, A.; Bhagavathula, A.S. Hypertriglyceridemia-Induced Acute Pancreatitis in a Patient with Type 2 Diabetes Mellitus. Cureus 2020, 12, e9414. [Google Scholar] [CrossRef] [PubMed]
- Lindkvist, B.; Appelros, S.; Regner, S.; Manjer, J. A prospective cohort study on risk of acute pancreatitis related to serum triglycerides, cholesterol and fasting glucose. Pancreatology 2012, 12, 317–324. [Google Scholar] [CrossRef]
- Bessembinders, K.; Wielders, J.; van de Wiel, A. Severe hypertriglyceridemia influenced by alcohol (SHIBA). Alcohol Alcohol 2011, 46, 113–116. [Google Scholar] [CrossRef] [Green Version]
- Zemánková, K.; Makoveichuk, E.; Vlasáková, Z.; Olivecrona, G.; Kovář, J. Acute alcohol consumption downregulates lipoprotein lipase activity in vivo. Metabolism 2015, 64, 1592–1596. [Google Scholar] [CrossRef]
- Esparza, M.I.; Li, X.; Adams-Huet, B.; Vasandani, C.; Vora, A.; Das, S.R.; Garg, A.; Ahmad, Z. Very Severe Hypertriglyceridemia in a Large US County Health Care System: Associated Conditions and Management. J. Endocr. Soc. 2019, 3, 1595–1607. [Google Scholar] [CrossRef]
- Simha, V. Management of hypertriglyceridemia. BMJ 2020, 371, m3109. [Google Scholar] [CrossRef] [PubMed]
- Chaggar, P.S.; Shaw, S.M.; Williams, S.G. Effect of antipsychotic medications on glucose and lipid levels. J. Clin. Pharmacol. 2011, 51, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Mehta, N.; Wayne, A.S.; Kim, Y.H.; Hale, G.A.; Alvarado, C.S.; Myskowski, P.; Jaffe, E.S.; Busam, K.J.; Pulitzer, M.; Zwerner, J.; et al. Bexarotene is active against subcutaneous panniculitis-like T-cell lymphoma in adult and pediatric populations. Clin. Lymphoma Myeloma Leuk. 2012, 12, 20–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javot, L.; Spaëth, D.; Scala-Bertola, J.; Gambier, N.; Petitpain, N.; Gillet, P. Severe hypertriglyceridaemia during treatment with capecitabine. Br. J. Cancer 2011, 104, 1238–1239. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.Q.; Li, W.X.; Zheng, J.J.; Tian, Q.-N.; Huang, J.-F.; Dai, S.-X. Gain and loss events in the evolution of the apolipoprotein family in vertebrata. BMC Evol. Biol. 2019, 19, 209. [Google Scholar] [CrossRef]
- O’Brien, P.J.; Alborn, W.E.; Sloan, J.H.; Ulmer, M.; Boodhoo, A.; Knierman, M.D.; Schultze, A.E.; Konrad, R.J. The novel apolipoprotein A5 is present in human serum, is associated with VLDL, HDL, and chylomicrons, and circulates at very low concentrations compared with other apolipoproteins. Clin. Chem. 2005, 51, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.; Forte, T.M.; Ryan, R.O. Influence of apolipoprotein A-V on the metabolic fate of triacylglycerol. Curr. Opin. Lipidol. 2013, 24, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Hubacek, J.A. Apolipoprotein A5 fifteen years anniversary: Lessons from genetic epidemiology. Gene 2016, 592, 193–199. [Google Scholar] [CrossRef]
- Dron, J.S.; Hegele, R.A. Genetics of Hypertriglyceridemia. Front. Endocrinol. (Lausanne) 2020, 11, 455. [Google Scholar] [CrossRef]
- Beckstead, J.A.; Oda, M.N.; Martin, D.D.; Forte, T.M.; Bielicki, J.K.; Berger, T.; Luty, R.; Kay, C.M.; Ryan, R.O. Structure-function studies of human apolipoprotein A-V: A regulator of plasma lipid homeostasis. Biochemistry 2003, 42, 9416–9423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forte, T.M.; Shu, X.; Ryan, R.O. The ins (cell) and outs (plasma) of apolipoprotein AV. J. Lipid Res. 2009, 50, S150–S155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, W.; Zhang, Z.; Yao, C.; Zhao, S. Emerging evidences for the opposite role of apolipoprotein C3 and apolipoprotein A5 in lipid metabolism and coronary artery disease. Lipids Health Dis. 2019, 18, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, R.; Stitziel, N.O.; Won, H.H.; Jørgensen, A.B.; Duga, S.; Angelica Merlini, P.; Kiezun, A.; Farrall, M.; Goel, A.; Zuk, O.; et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature 2015, 518, 102–106. [Google Scholar] [CrossRef]
- Dussaillant, C.; Serrano, V.; Maiz, A.; Eyheramendy, S.; Cataldo, L.R.; Chavez, M.; Smalley, S.V.; Fuentes, M.; Rigotti, A.; Rubio, L.; et al. APOA5 Q97X mutation identified through homozygosity mapping causes severe hypertriglyceridemia in a Chilean consanguineous family. BMC Med. Genet. 2012, 13, 106. [Google Scholar] [CrossRef] [Green Version]
- Jasim, A.A.; Al-Bustan, S.A.; Al-Kandari, W.; Al-Serri, A.; AlAskar, H. Sequence Analysis of APOA5 Among the Kuwaiti Population Identifies Association of rs2072560, rs2266788, and rs662799 With TG and VLDL Levels. Front. Genet. 2018, 9, 112. [Google Scholar] [CrossRef] [Green Version]
- Oliva, P.; Carubbi, F.; Schaap, F.G.; Bertolini, S.; Calandra, S. Hypertriglyceridemia and low plasma HDL in a patient with apolipoprotein A-V deficiency due to a novel mutation in the APOA5 gene. J. Intern. Med. 2008, 263, 450–458. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Total Cholesterol | LDL Cholesterol | HDL Cholesterol | Triglycerides | Notes | |
|---|---|---|---|---|---|
| Normal range | 3.2–5.2 | 1.6–3.8 | 1.1–2.3 | 0.1–1.7 | |
| Mother, 36 years old | 4.1 | 2.4 | 1.3 | 0.8 | — |
| Father, 34 years old | 4.0 | 1.0 | 0.7 | 6.8 | Plasma lactescence |
| Brother, 11 years old | — | — | — | 2.0 | — |
| Uncle (p), 30 years old | 4.0 | 1.5 | 0.5 | 5.3 | — |
| Aunt (p), 40 years old | 5.9 | 2.0 | 0.9 | 7.7 | — |
| Date | Total Cholesterol | LDL Cholesterol | HDL Cholesterol | Triglycerides | Notes |
|---|---|---|---|---|---|
| Normal range | 3.2–5.2 | 1.6–3.8 | 1.1–2.3 | 0.1–1.7 | |
| 13 January 2017 | 11.2 | — | 0.5 | 55.1 | Plasma lactescence |
| 7 February 2017 | 9.4 | — | — | 16.1 | Plasma lactescence |
| 17 August 2017 | 3.9 | 1.2 | 0.5 | 6.3 | Plasma lactescence |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasiluev, P.A.; Ivanova, O.N.; Semenova, N.A.; Strokova, T.V.; Taran, N.N.; Chubykina, U.V.; Ezhov, M.V.; Zakharova, E.Y.; Dadli, E.L.; Kutsev, S.I. A Clinical Case of a Homozygous Deletion in the APOA5 Gene with Severe Hypertriglyceridemia. Genes 2022, 13, 1062. https://doi.org/10.3390/genes13061062
Vasiluev PA, Ivanova ON, Semenova NA, Strokova TV, Taran NN, Chubykina UV, Ezhov MV, Zakharova EY, Dadli EL, Kutsev SI. A Clinical Case of a Homozygous Deletion in the APOA5 Gene with Severe Hypertriglyceridemia. Genes. 2022; 13(6):1062. https://doi.org/10.3390/genes13061062
Chicago/Turabian StyleVasiluev, Petr Andreevich, Olga N. Ivanova, Natalia A. Semenova, Tatiana V. Strokova, Natalia N. Taran, Uliana V. Chubykina, Marat V. Ezhov, Ekaterina Y. Zakharova, Elena L. Dadli, and Sergey I. Kutsev. 2022. "A Clinical Case of a Homozygous Deletion in the APOA5 Gene with Severe Hypertriglyceridemia" Genes 13, no. 6: 1062. https://doi.org/10.3390/genes13061062