
Genetic Profile of Patients with Limb-Girdle Muscle Weakness in the Chilean Population

 ,
,  , , , , , , , ,
, , , , , , , ,
Abstract
:1. Introduction
2. Patients and Methods
2.1. Patient Enrollment
2.2. Sequencing
- MyoPanel2, consisting of 306 neuromuscular disease-causing genes designed at the Marseille Medical Genetics Institute (Aix-Marseille University, Marseille, France) [14]. The enrichment was performed using HaloPlex technology (Agilent TechnologiesTM), followed by sequencing on the NextSeq500 (IlluminaTM) by HelixioTM (Biopôle Clermont-Limagne, France). The bioinformatic analysis was performed as previously described [14];
- DLE-NGS—DLE Laboratory, Sao Paulo, Brazil, consisting of ten genes, the nine most-frequent LGMD-causing genes: CAPN3; DYSF; SGCG; SGCA; SGCB; SGCD; FKRP; ANO5; TCAP and GAA, as described elsewhere [10]. The coding regions and 10 nucleotides from the exon-intron junction from the included genes and intronic variants were customized with Agilent Sure-Select capture covering above 98% of target regions at 20x or greater. Nine genes and 154 corresponding exons related to muscular dystrophies and Pompe disease were included. Deep intronic variants were also investigated. Flanking exon/intron regions up to 25 base pairs (bp) were sequenced as well as known intronic variants if outside this range. The coding and flanking intronic regions were enriched using a Custom SureSelect QXT kit (Agilent technology) and were sequenced using the Illumina NextSeq 500 system. Only variants (SNVs/small indels) in the coding region and the flanking intronic regions (+10 bp) with a minor allele frequency (MAF) < 5% were evaluated. The ExAC, 1000Genomes and ABraOM projects were used to determine the frequency of the variants; a CADD score over 20 was the threshold to classify the in silico damaging prediction of the variant to the final protein, and other published information and laboratory databanks were used to further classify the variants. Patients who had pathogenic variants in homozygous or compound heterozygous state for GAA consistent with Pompe disease had alpha-glucosidase activity measured in the same paper filter card by fluorometry. After sequencing, the base call generated BCL files that were converted to FASTQ using the BCL2FASTQ script. The aligned file was then used for calling variants with the Samtools software, followed by annotation using the Variant Effect Predictor (VEP). “VCF” files annotated with VEP and in-house scripts were converted to tabulated tables and incorporated frequency information from variants already sequenced as well as Reactome and OMIM information. Quality analysis of the sequencing and call of variants was conducted by FASTQ and BAM files checked with Qualimap software. In addition, the average size of sequenced reads, aligned reads, transition rate, transversion, insertion, and deletion were surveyed. The nomenclature followed the HGVS guidelines [10];
- CL-NGS panel, set-up at the Instituto de Ciencias Biomédicas, Facultad de Medicina, Universidad de Chile, comprising 15 neuromuscular disease-associated genes including LMNA; CAV3; DNAJB6; CAPN3; DYSF; SGCG; SGCA; SGCB; SGCD; FKRP; ANO5; FKTN; EMD; FHL1 and DES. Enrichment of coding regions by multiplex PCR and library preparation was performed with the Ion AmpliSeq Library Kit 2.0 (Thermo Fisher Scientific Inc., Waltham, MA, USA) according to instructions by the manufacturer. The pool of primers used for multiplex PCR was designed with the Ion Ampliseq Designer 5.6.3 tool (Thermo Fisher Scientific) covering 99% of the target regions. It amplified the targeted exons and >10 bp of surrounding intronic regions, with amplicons ranging from 125–375 bp. Emulsion PCR for clonal amplification of DNA in spheres was performed with the Ion PGM OT2 Hi-Q view kit (Thermo Fisher Scientific) on OneTouch 2 equipment. Sequencing was performed on an Ion Personal Genome Machine (Ion PGM system, Applied Biosystems) sequencer with the Ion sequencing kit, PGM Hi-Q view (Thermo Fisher Scientific), using the protocol provided by the manufacturer. The number of samples to be sequenced per run was calculated to achieve a minimum coverage depth of 100×. The variant calling was made with the Ion Reporter software (Thermo Fisher Scientific) using the default germline variant settings and hg19 as the genome reference;
- Invitae Comprehensive Neuromuscular Disorders Panel (CNMDP), comprised of 123 neuromuscular disease-causative genes including deletions and duplications (https://www.invitae.com/en/providers/test-catalog/test-03280, accessed on 1 April 2019). For the case of P53-Myo120, a panel of 137 genes was used. Genomic DNA obtained from the sample was enriched for targeted regions using a hybridization-based protocol and sequenced using Illumina technology. All targeted regions were sequenced with ≥50× depth or supplemented with additional analysis. Reads were aligned to a reference sequence (GRCh37/hg19), and sequence changes were identified and interpreted in the context of a single clinically relevant transcript. Enrichment and analysis focused on the coding sequence of the indicated transcripts, 20 bp of flanking intronic sequence and other specific genomic regions demonstrated to be causative of disease at the time of assay design. Promoters, untranslated regions and other noncoding regions were not otherwise interrogated. For some genes only, targeted loci were analyzed (Supplementary Materials File S1). Exonic deletions and duplications were called using an in-house algorithm that determined the copy number at each target by comparing the read depth for each target in the proband sequence with both mean read-depth and read-depth distribution, obtained from a set of clinical samples. TTN exons 45-46, 147, 149, 164 and 172-201 (NM_001267550.2) were excluded from analysis. This assay unambiguously detects SMN1 exon 8 copy number and sequence variants as well as sequence variants outside of exon 8, but this assay cannot determine whether the variant is in SMN1 or SMN2. CNVs of exons 1–6 of SMN1 or SMN2 are not reported. Confirmation technologies included any of the following: Sanger sequencing, Pacific Biosciences SMRT sequencing, MLPA and Array CGH.
2.3. DNA Extraction and Sample Collection
2.4. Variant Analysis
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cohen, E.; Bonne, G.; Rivier, F.; Hamroun, D. The 2022 version of the gene table of neuromuscular disorders. Neuromuscul. Disord. 2021, 31, 1313–1357. [Google Scholar] [CrossRef] [PubMed]
- Monges, S.; Rosa, A. Management of neuromuscular diseases and spinal muscular atrophy in Latin America. Gene Ther. 2017, 24, 578–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cea, J.G. Aproximación a las Miopatías Hereditarias. In Tratado de Neurología Clínica, 1st ed.; Nogales-Gaete, J., Donoso, A., Verdugo, R., Eds.; Editorial Universitaria: Santiago, Chile, 2005; ISBN 9561117983. [Google Scholar]
- Wicklund, M.P. The limb-girdle muscular dystrophies. CONTINUUM Lifelong Learn. Neurol. 2019, 25, 1599–1618. [Google Scholar] [CrossRef] [PubMed]
- Georganopoulou, D.G.; Moisiadis, V.G.; Malik, F.A.; Mohajer, A.; Dashevsky, T.M.; Wuu, S.T.; Hu, C.K. A Journey with LGMD: From Protein Abnormalities to Patient Impact. Protein J. 2021, 40, 466–488. [Google Scholar] [CrossRef] [PubMed]
- Straub, V.; Murphy, A.; Udd, B. LGMD Workshop Study. 229th ENMC international workshop: Limb girdle muscular dystrophies—Nomenclature and reformed classification Naarden, the Netherlands, 17–19 March 2017. Neuromuscul. Disord. 2018, 28, 702–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmood, O.A.; Jiang, X.M. Limb-girdle muscular dystrophies: Where next after six decades from the first proposal (Review). Mol. Med. Rep. 2014, 9, 1515–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Pajusalu, S.; Lake, N.J.; Zhou, G.; Ioannidis, N.; Mittal, P.; Johnson, N.E.; Weihl, C.C.; Williams, B.A.; Albrecht, D.E.; et al. Estimating prevalence for limb-girdle muscular dystrophy based on public sequencing databases. Genet Med. 2019, 21, 2512–2520. [Google Scholar] [CrossRef] [PubMed]
- Nallamilli, B.R.R.; Chakravorty, S.; Kesari, A.; Tanner, A.; Ankala, A.; Schneider, T.; da Silva, C.; Beadling, R.; Alexander, J.J.; Askree, S.H.; et al. Genetic landscape and novel disease mechanisms from a large LGMD cohort of 4656 patients. Ann. Clin. Transl. Neurol. 2018, 5, 1574–1587. [Google Scholar] [CrossRef] [PubMed]
- Bevilacqua, J.A.; Guecaimburu-Ehuletche, M.D.R.; Perna, A.; Dubrovsky, A.; Franca, M.C.; Vargas, S.; Hegde, M.; Claeys, K.G.; Straub, V.; Daba, N.; et al. The Latin American experience with a next generation sequencing genetic panel for recessive limb-girdle muscular weakness and Pompe disease. Orphanet J. Rare Dis. 2020, 15, 11. [Google Scholar] [CrossRef] [Green Version]
- Töpf, A.; Johnson, K.; Bates, A.; Phillips, L.; Chao, K.R.; England, E.M.; Laricchia, K.M.; Mullen, T.; Valkanas, E.; Xu, L.; et al. Sequential targeted exome sequencing of 1001 patients affected by unexplained limb girdle weakness. Genet. Med. 2020, 22, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- González-Quereda, L.; Rodríguez, M.J.; Díaz-Manera, J.; Alonso-Pérez, J.; Gallardo, E.; Nascimento, A.; Ortez, C.; Natera-de Benito, D.; Olivé, M.; González-Mera, L.; et al. Targeted Next-Generation Sequencing in a Large Cohort of Genetically Undiagnosed Patients with Neuromuscular Disorders in Spain. Genes 2020, 11, 539. [Google Scholar] [CrossRef]
- Savarese, M.; Di Fruscio, G.; Mutarelli, M.; Torella, A.; Magri, F.; Santorelli, F.M.; Comi, G.P.; Bruno, C.; Nigro, V. MotorPlex provides accurate variant detection across large muscle genes both in single myopathic patients and in pools of DNA samples. Acta Neuropathol. Commun. 2014, 2, 100. [Google Scholar] [CrossRef]
- Gorokhova, S.; Cerino, M.; Mathieu, Y.; Courrier, S.; Desvignes, J.P.; Salgado, D.; Béroud, C.; Krahn, M.; Bartoli, M. Comparing targeted exome and whole exome approaches for genetic diagnosis of neuromuscular disorders. Appl. Transl. Genom. 2015, 7, 26–31. [Google Scholar] [CrossRef]
- Kuhn, M.; Glaser, D.; Joshi, P.R.; Zierz, S.; Wenninger, S.; Schoser, B.; Deschauer, M. Utility of a next-generation sequencing-based gene panel investigation in German patients with genetically unclassified limb-girdle muscular dystrophy. J. Neurol. 2016, 263, 743–750. [Google Scholar] [CrossRef]
- Savarese, M.; Di Fruscio, G.; Torella, A.; Fiorillo, C.; Magri, F.; Fanin, M.; Ruggiero, L.; Ricci, G.; Astrea, G.; Passamano, L.; et al. The genetic basis of undiagnosed muscular dystrophies and myopathies: Results from 504 patients. Neurology 2016, 87, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Yubero, D.; Natera-de Benito, D.; Pijuan, J.; Armstrong, J.; Martorell, L.; Fernández, G.; Maynou, J.; Jou, C.; Roldan, M.; Ortez, C.; et al. The Increasing Impact of Translational Research in the Molecular Diagnostics of Neuromuscular Diseases. Int. J. Mol. Sci. 2021, 22, 4274. [Google Scholar] [CrossRef]
- Bevilacqua, J.A.; Lara, M.; Díaz, J.; Campero, M.; Vázquez, J.; Maselli, R.A. Congenital Myasthenic Syndrome due to DOK7 mutations in a family from Chile. Eur. J. Transl. Myol. 2017, 27, 6832. [Google Scholar] [CrossRef] [Green Version]
- Bevilacqua, J.A.; González-Quereda, L.; Castiglioni, C.; Zamorano, I.; Acevedo, L.; Díaz, J.; Rodríguez, M.; Trangulao, A.; Rivera, M.; Gallano, P. Desminopathy in Chile, first cases reported. Neuromuscul. Disord. 2016, 26 (Suppl. S114), 106. [Google Scholar] [CrossRef]
- González-Quereda, L.; Fuentealba, M.; Díaz, J.; Trangulao, A.; Gallano, P.; Bevilacqua, J.A. Distal upper limb onset myopathy in the first Chilean case reported with titinopathy. Neuromuscul. Disord. 2018, 28 (Suppl. S104), 288. [Google Scholar] [CrossRef]
- Richard, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Charnay, T.; Blanck, V.; Cerino, M.; Bartoli, M.; Riccardi, F.; Bonello-Palot, N.; Pécheux, C.; Nguyen, K.; Lévy, N.; Gorokhova, S.; et al. Retrospective analysis and reclassification of DYSF variants in a large French series of dysferlinopathy patients. Genet. Med. 2021, 23, 1574–1577. [Google Scholar] [CrossRef]
- Savarese, M.; Johari, M.; Johnson, K.; Arumilli, M.; Torella, A.; Töpf, A.; Rubegni, A.; Kuhn, M.; Giugliano, T.; Gläser, D.; et al. Improved Criteria for the Classification of Titin Variants in Inherited Skeletal Myopathies. J. Neuromuscul. Dis. 2020, 7, 153–166. [Google Scholar] [CrossRef]
- Abou Tayoun, A.N.; Pesaran, T.; DiStefano, M.T.; Oza, A.; Rehm, H.L.; Biesecker, L.G.; Harrison, S.M. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 2018, 39, 1517–1524. [Google Scholar] [CrossRef]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [Green Version]
- Desmet, F.-O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [Green Version]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput Biol. J. Comput. Mol. Cell Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef] [Green Version]
- Oza, A.M.; DiStefano, M.T.; Hemphill, S.E.; Cushman, B.J.; Grant, A.R.; Siegert, R.K.; Shen, J.; Chapin, A.; Boczek, N.J.; Schimmenti, L.A.; et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 2018, 39, 1593–1613. [Google Scholar] [CrossRef] [PubMed]
- Whiffin, N.; Minikel, E.; Walsh, R.; O’Donnell-Luria, A.H.; Karczewski, K.; Ing, A.Y.; Barton, P.J.R.; Funke, B.; Cook, S.A.; MacArthur, D.; et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet. Med. J. Am. Coll. Med. Genet. 2017, 19, 1151–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biesecker, L.G.; Harrison, S.M. ClinGen Sequence Variant Interpretation Working Group. The ACMG/AMP reputable source criteria for the interpretation of sequence variants. Genet. Med. J. Am. Coll. Med. Genet. 2018, 20, 1687–1688. [Google Scholar] [CrossRef]
- Bevilacqua, J.A.; Mathieu, Y.; Krahn, M.; Bartoli, M.; Castiglioni, C.; Kleinsteuber, K.; Díaz, J.; Puppo, F.; Cerino, M.; Courrier, S.; et al. Calpainopathy in Chile, first Cases reports. Neuromuscul. Disord. 2016, 26 (Suppl. S91), 23. [Google Scholar] [CrossRef]
- Bevilacqua, J.A.; Contreras, J.P.; Trangulao, A.; Hernández, U.M.; Brochier, G.; Diaz, J.; Hughes, R.; Campero, M.; Romero, N.B. Novel autosomal dominant TPM3 mutation causes a combined congenital fibre type disproportion-cap disease histological pattern. Neuromuscul. Disord. 2022, in press. [Google Scholar] [CrossRef]
- Savarese, M.; Sarparanta, J.; Vihola, A.; Udd, B.; Hackman, P. Increasing Role of Titin Mutations in Neuromuscular Disorders. J. Neuromuscul. Dis. 2016, 3, 293–308. [Google Scholar] [CrossRef] [Green Version]
- Schiava, M.; Marchesoni, C.; García de Rosa, M.L.; Estrada, N.; Cejas, L.L.; Pardal, A.; Pirra, L.; Repetto, L.; Torres, A.; Dubrovsky, A.; et al. Genetic characterization of Limb Girdle Muscular Dystrophies and Pompe Disease in a large Argentine cohort. Neurol. Perspect. 2022, in press. [Google Scholar] [CrossRef]
- Winckler, P.B.; Chwal, B.C.; Dos Santos, M.A.R.; Burguêz, D.; Polese-Bonatto, M.; Zanoteli, E.; Siebert, M.; Vairo, F.P.E.; Chaves, M.L.F.; Saute, J.A.M. Diagnostic yield of multi-gene panel for muscular dystrophies and other hereditary myopathies. Neurol. Sci. 2022, 17. [Google Scholar] [CrossRef]
- Yu, M.; Zheng, Y.; Jin, S.; Gang, Q.; Wang, Q.; Yu, P.; Lv, H.; Zhang, W.; Yuan, Y.; Wang, Z. Mutational spectrum of Chinese LGMD patients by targeted next-generation sequencing. PLoS ONE 2017, 12, e0175343. [Google Scholar] [CrossRef] [Green Version]
- Suárez, B.; Jofré, J.; Lozano-Arango, A.; Ortega, X.; Díaz, J.; Calcagno, G.; Bevilacqua, J.A.; Castiglioni, C. Spontaneous symptomatic improvement in a pediatric patient with anti-3-hydroxy-3-methylglutraryl-coenzyme A reductase myopathy. Neuromuscul. Disord. 2020, 30, 503–509. [Google Scholar] [CrossRef]
- Mohassel, P.; Landon-Cardinal, O.; Reghan Foley, A.; Donkervoort, S.; Pak, K.S.; Wahl, C.; Shebert, R.T.; Harper, A.; Fequiere, P.; Meriggioli, M.; et al. Anti-HMGCR myopathy may resemble limb-girdle muscular dystrophy. Neurol. Neuroimmunol. Neuroinflamm. 2019, 6, e523. [Google Scholar] [CrossRef] [Green Version]
- Mammen, A.L. Autoimmune myopathies: Autoantibodies, phenotypes and pathogenesis. Nat. Rev. Neurol. 2011, 7, 343–354. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Ginjaar, I.B.; Bushby, K. The importance of genetic diagnosis for Duchenne muscular dystrophy. J. Med. Genet. 2016, 53, 145–151. [Google Scholar] [CrossRef]
- Fernández-Eulate, G.; Querin, G.; Moore, U.; Behin, A.; Masingue, M.; Bassez, G.; Leonard-Louis, S.; Laforêt, P.; Maisonobe, T.; Merle, P.E.; et al. Deep phenotyping of an international series of patients with late onset dysferlinopathy. Eur. J. Neurol. 2021, 28, 2092–2102. [Google Scholar] [CrossRef] [PubMed]
- Woudt, L.; Di Capua, G.A.; Krahn, M.; Castiglioni, C.; Hughes, R.; Campero, M.; Trangulao, A.; González-Hormazábal, P.; Godoy-Herrera, R.; Levy, N.; et al. Toward an objective measure of functional disability in dysferlinopathy. Muscle Nerve 2016, 53, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Andrés, D.; Díaz, J.; Munell, F.; Sánchez-Montáñez, A.; Pulido-Valdeolivas, I.; Suazo, L.; Garrido, C.; Quijano-Roy, S.; Bevilacqua, J.A. Disease duration and disability in dysferlinopathy can be described by muscle imaging using heatmaps and random forests. Muscle Nerve 2019, 59, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Reghan Foley, A.; Donkervoort, S.; Bonnemann, C.G. Next generation sequencing still needs our generation’s clinicians. Neurol. Genet. 2015, 1, e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Medical Association Declaration of Helsinki: Ethical principles for medical research involving human subjects. JAMA 2013, 310, 2191–2194. [CrossRef] [PubMed] [Green Version]

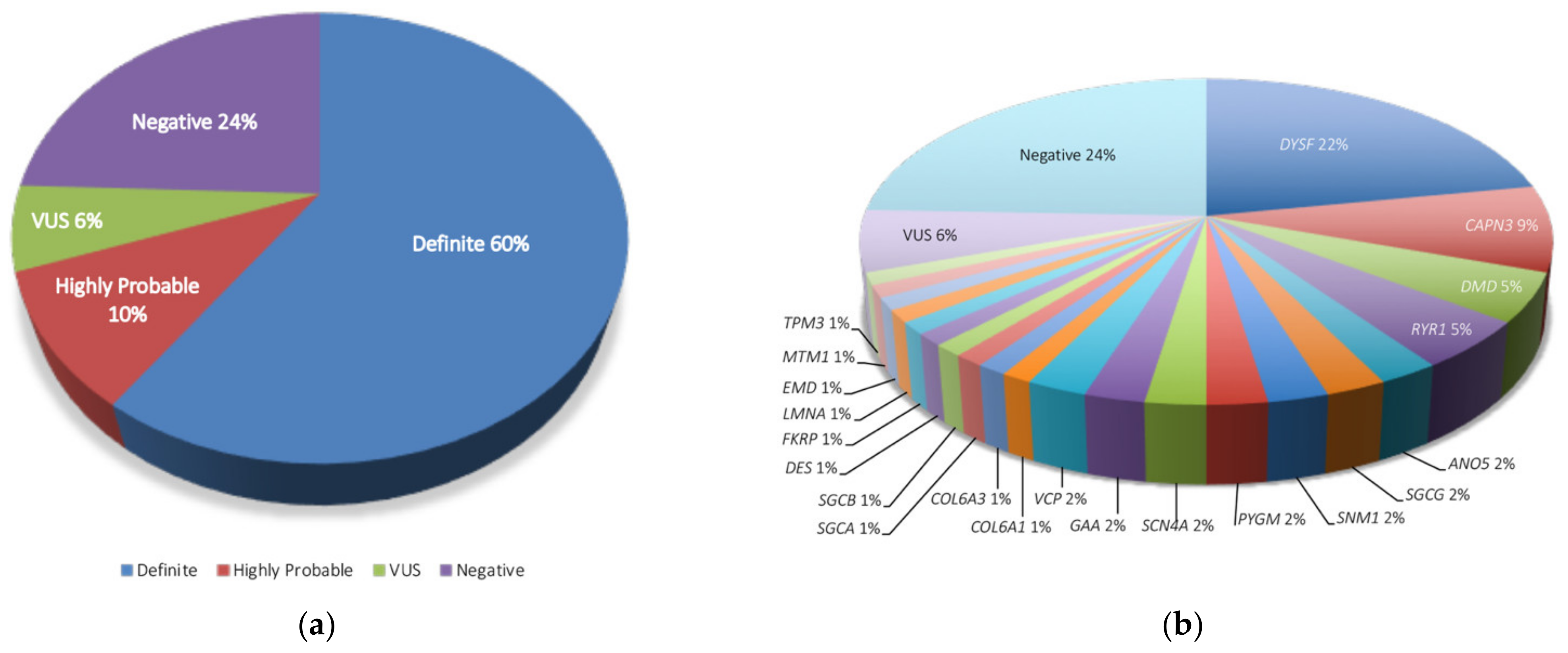

{kind=link}
{kind=link}
| Parameter | Statistics |
|---|---|
| Total, n | 82 |
| Female/male, n (%) | 35 (42.7)/47 (57.3) |
| Age (y), mean ± SD (minimum–maximum) | 36.8 ± 13.9 (8–68) |
| <18 years of age, n (%) | 7 (8.5) |
| ≥18 years of age, n (%) | 75 (91.5) |
| Patients with definite genetic diagnosis, n (%) | 49 (59.8) |
| Patients with highly probable genetic diagnosis, n (%) | 8 (9.8) |
| Patients without genetic diagnosis, n (%) | 25 (30.5) |
| Patients with any * conclusive diagnosis, n (%) | 63 (76.8) |
| Patients without conclusive diagnosis, n (%) | 19 (23.2) |
| Time from onset of diagnosis (y), mean ± SD (minimum–maximum) | 11.2 ± 11.3 (3–50) |
| Geographic Origin ** | n (%) |
| Northern Chile | 7 (8.5) |
| Central Chile | 6 (7.3) |
| Southern Chile | 13 (15.9) |
| Santiago Metropolitan Area | 53 (64.6) |
| Ecuador | 1 (1.2) |
| Bolivia | 1 (1.2) |
| Peru | 1 (1.2) |
| Gene | n, Affected Patients (% over 82 Samples) |
|---|---|
| DYSF | 18 (22) |
| CAPN3 | 7 (8.5) |
| DMD | 4 (4.9) |
| RYR1 | |
| ANO5 | 2 (2.4) |
| SGCG | |
| SNM1 | |
| PGYM | |
| SCN4A | |
| GAA | |
| VCP | |
| COL6A1 | 1 (1.2) |
| COL6A3 | |
| SGCA | |
| SGCB | |
| DES | |
| FKRP | |
| LMNA | |
| EMD | |
| MTM1 | |
| TPM3 | |
| VUS | 5 (6.1) |
| Negative | 20 (24.4) |
| Total | 82 (100) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cerino, M.; González-Hormazábal, P.; Abaji, M.; Courrier, S.; Puppo, F.; Mathieu, Y.; Trangulao, A.; Earle, N.; Castiglioni, C.; Díaz, J.; et al. Genetic Profile of Patients with Limb-Girdle Muscle Weakness in the Chilean Population. Genes 2022, 13, 1076. https://doi.org/10.3390/genes13061076
Cerino M, González-Hormazábal P, Abaji M, Courrier S, Puppo F, Mathieu Y, Trangulao A, Earle N, Castiglioni C, Díaz J, et al. Genetic Profile of Patients with Limb-Girdle Muscle Weakness in the Chilean Population. Genes. 2022; 13(6):1076. https://doi.org/10.3390/genes13061076
Chicago/Turabian StyleCerino, Mathieu, Patricio González-Hormazábal, Mario Abaji, Sebastien Courrier, Francesca Puppo, Yves Mathieu, Alejandra Trangulao, Nicholas Earle, Claudia Castiglioni, Jorge Díaz, and et al. 2022. "Genetic Profile of Patients with Limb-Girdle Muscle Weakness in the Chilean Population" Genes 13, no. 6: 1076. https://doi.org/10.3390/genes13061076
APA StyleCerino, M., González-Hormazábal, P., Abaji, M., Courrier, S., Puppo, F., Mathieu, Y., Trangulao, A., Earle, N., Castiglioni, C., Díaz, J., Campero, M., Hughes, R., Vargas, C., Cortés, R., Kleinsteuber, K., Acosta, I., Urtizberea, J. A., Lévy, N., Bartoli, M., ... Bevilacqua, J. A. (2022). Genetic Profile of Patients with Limb-Girdle Muscle Weakness in the Chilean Population. Genes, 13(6), 1076. https://doi.org/10.3390/genes13061076