
Cellular Senescence in Normal Mammary Gland and Breast Cancer. Implications for Cancer Therapy

 and
and {kind=link}
{kind=link}
Abstract
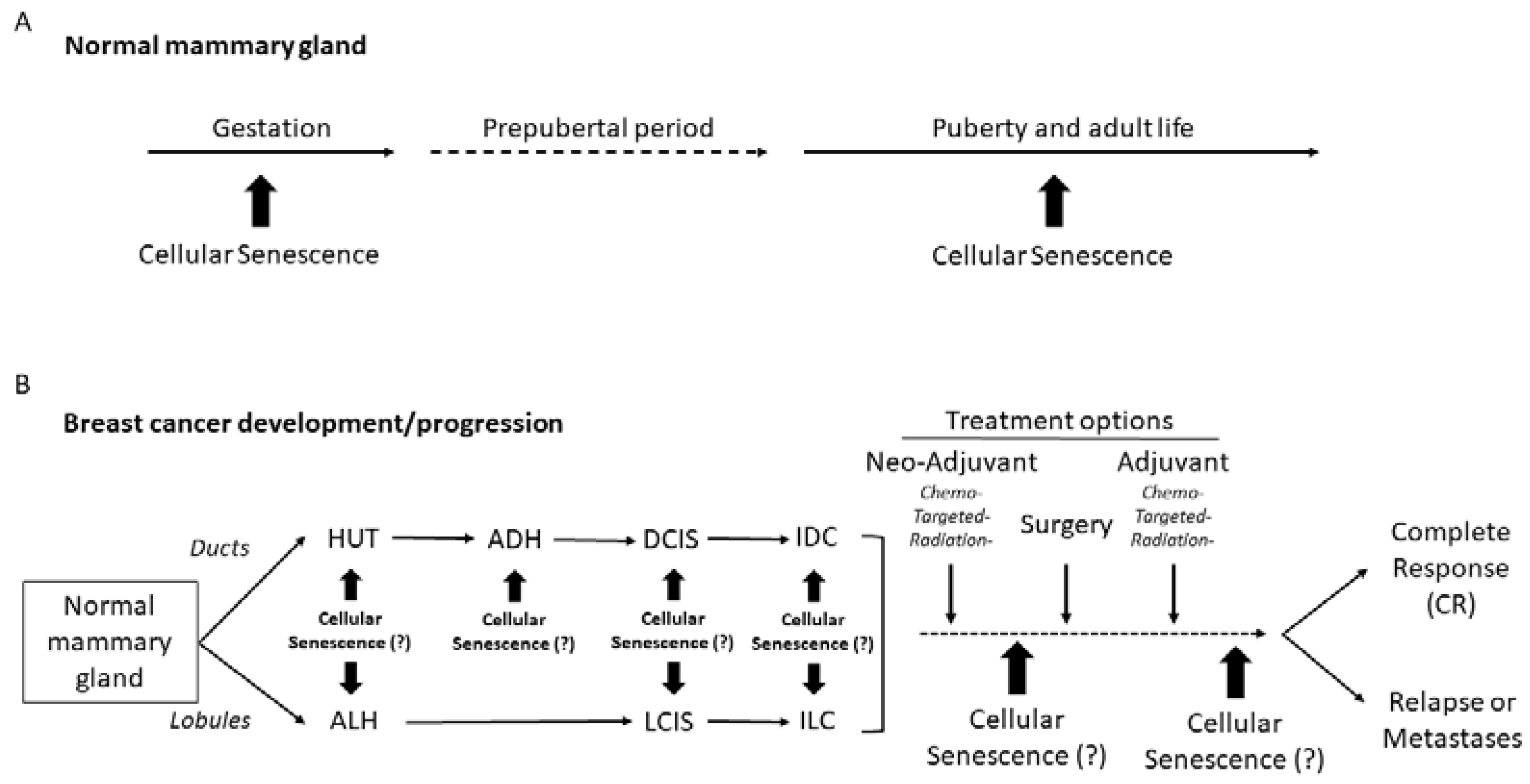
:1. Introduction
2. Senescence in Normal and Non-Tumorigenic Human Mammary Epithelial Cells
2.1. Paradigm of Normal Human Mammary Epithelial Cells (HMECs)
2.2. The Paradigm of the MCF10A Non-Tumorigenic Mammary Cell Line
3. Cellular Senescence in Breast Carcinoma
4. Cellular Senescence and Breast-Cancer Treatment
5. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Rosen, P.P. Anatomy and physiological morphology. In Rosen’s Breast Pathology; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2009; pp. 1–25. [Google Scholar]
- Marchio, C.; Geyer, F.C.; Reis-Filho, J.S. Pathology and Molecular Pathology of the Breast. In Pathology and Epidemiology of Cancer; Loda, M., Mucci, L.A., Mittelstadt, M.L., van Hemelrijck, M., Cotter, M.B., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 173–231. [Google Scholar]
- Ham, A.W.; Comrack, D.H. The breast. In Histology; Lipponcott: Philadelphia, PA, USA, 1979; pp. 866–874. [Google Scholar]
- Adriance, M.C.; Inman, J.L.; Petersen, O.W.; Bissell, M.J. Myoepithelial cells: Good fences make good neighbors. Breast Cancer Res. 2005, 7, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Topper, Y.J.; Freeman, C.S. Multiple hormone interactions in the developmental biology of the mammary gland. Physiol. Rev. 1980, 60, 1049–1106. [Google Scholar] [CrossRef]
- Sternlicht, M.D. Key stages in mammary gland development: The cues that regulate ductal branching morphogenesis. Breast Cancer Res. 2005, 8, 1. [Google Scholar] [CrossRef] [Green Version]
- Bartkova, J.; Hořejší, Z.; Koed, K.; Krämer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef]
- Bermejo, R.; Lai, M.S.; Foiani, M. Preventing replication stress to maintain genome stability: Resolving conflicts between replication and transcription. Mol. Cell 2012, 45, 710–718. [Google Scholar] [CrossRef] [Green Version]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Mazouzi, A.; Velimezi, G.; Loizou, J.I. DNA replication stress: Causes, resolution and disease. Exp. Cell Res. 2014, 329, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Giovannelli, P.; Di Donato, M.; Galasso, G.; Di Zazzo, E.; Bilancio, A.; Migliaccio, A. The androgen receptor in breast cancer. Front. Endocrinol. 2018, 9, 492. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Garcia, M.A.; Geyer, F.C.; Lacroix-Triki, M.; Marchió, C.; Reis-Filho, J.S. Breast cancer precursors revisited: Molecular features and progression pathways. Histopathology 2010, 57, 171–192. [Google Scholar] [CrossRef]
- Humphreys, R.C.; Krajewska, M.; Krnacik, S.; Jæger, R.; Weiher, H.; Krajewski, S.; Reed, J.C.; Rosen, J.M. Apoptosis in the terminal endbud of the murine mammary gland: A mechanism of ductal morphogenesis. Development 1996, 122, 4013–4022. [Google Scholar] [CrossRef] [PubMed]
- Mailleux, A.A.; Overholtzer, M.; Schmelzle, T.; Bouillet, P.; Strasser, A.; Brugge, J.S. BIM regulates apoptosis during mammary ductal morphogenesis, and its absence reveals alternative cell death mechanisms. Dev. Cell 2007, 12, 221–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, H.C.; Avivar-Valderas, A.; Sosa, M.S.; Girnius, N.; Farias, E.F.; Davis, R.J.; Aguirre-Ghiso, J.A. p38α signaling induces anoikis and lumen formation during mammary morphogenesis. Sci. Signal. 2011, 4, ra34. [Google Scholar] [CrossRef] [Green Version]
- Avivar-Valderas, A.; Salas, E.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Nagi, C.; Debnath, J.; Aguirre-Ghiso, J.A. PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Mol. Cell. Biol. 2011, 31, 3616–3629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivaraman, L.; Conneely, O.M.; Medina, D.; O’Malley, B.W. p53 is a potential mediator of pregnancy and hormone-induced re-sistance to mammary carcinogenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 12379–12384. [Google Scholar] [CrossRef] [Green Version]
- Debnath, J.; Mills, K.R.; Collins, N.L.; Reginato, M.J.; Muthuswamy, S.K.; Brugge, J.S. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell 2002, 111, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Karantza-Wadsworth, V.; Patel, S.; Kravchuk, O.; Chen, G.; Mathew, R.; Jin, S.; White, E. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007, 21, 1621–1635. [Google Scholar] [CrossRef] [Green Version]
- Lyons, T.R.; O’brien, J.; Borges, V.F.; Conklin, M.W.; Keely, P.J.; Eliceiri, K.W.; Marusyk, A.; Tan, A.C.; Schedin, P. Postpartum mammary gland involution drives progression of ductal carcinoma in situ through collagen and COX-2. Nat. Med. 2011, 17, 1109–1115. [Google Scholar] [CrossRef] [Green Version]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Gorgoulis, V.G.; Halazonetis, T.D. Oncogene-induced senescence: The bright and dark side of the response. Curr. Opin. Cell Biol. 2010, 22, 816–827. [Google Scholar] [CrossRef]
- He, S.; Sharpless, N.E. Senescence in health and disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. 249. Genes Dev. 2010, 24, 250. [Google Scholar] [CrossRef] [Green Version]
- Shay, J.W. Role of telomeres and telomerase in aging and cancer. Cancer Discov. 2016, 6, 584–593. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Schmitt, C.A. The dynamic nature of senescence in cancer. Nat. Cell Biol. 2019, 21, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Brenner, A.J.; Aldaz, C.M. Chromosome 9p allelic loss and p16/CDKN2 in breast cancer and evidence of p16 inactivation in immortal breast epithelial cells. Cancer Res. 1995, 55, 2892–2895. [Google Scholar]
- Brenner, A.J.; Stampfer, M.R.; Aldaz, C.M. Increased p16 expression with first senescence arrest in human mammary epithelial cells and extended growth capacity with p16 inactivation. Oncogene 1998, 17, 199–205. [Google Scholar] [CrossRef] [Green Version]
- Bean, G.R.; Bryson, A.D.; Pilie, P.G.; Goldenberg, V.; Baker, J.C.; Ibarra, C.; Brander, D.M.; Paisie, C.; Case, N.R.; Gauthier, M.; et al. Morphologically normal-appearing mammary epithelial cells obtained from high-risk women exhibit methylation silencing of INK4a/ARF. Clin. Cancer Res. 2007, 13, 6834–6841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romanov, S.R.; Kozakiewicz, B.K.; Holst, C.R.; Stampfer, M.R.; Haupt, L.M.; Tlsty, T.D. Normal human mammary epithelial cells spontaneously escape senescence and acquire genomic changes. Nature 2001, 409, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Tlsty, T.D.; Romanov, S.R.; Kozakiewicz, B.K.; Holst, C.R.; Haupt, L.M.; Crawford, Y.G. Loss of chromosomal integrity in human mammary epithelial cells subsequent to escape from senescence. J. Mammary Gland. Biol. Neoplasia 2001, 6, 235–243. [Google Scholar] [CrossRef]
- Berman, H.K.; Gauthier, M.L.; Tlsty, T.D. Premalignant breast neoplasia: A paradigm of interlesional and intralesional molecular heterogeneity and its biological and clinical ramifications. Cancer Prev. Res. 2010, 3, 579–587. [Google Scholar] [CrossRef] [Green Version]
- Crawford, Y.G.; Gauthier, M.L.; Joubel, A.; Mantei, K.; Kozakiewicz, K.; Afshari, C.A.; Tlsty, T.D. Histologically normal human mammary epithelia with silenced p16INK4a overexpress COX-2, promoting a premalignant program. Cancer Cell 2004, 5, 263–273. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Pickering, C.R.; Holst, C.R.; Gauthier, M.L.; Tlsty, T.D. p16INK4a modulates p53 in primary human mammary epithelial cells. Cancer Res. 2006, 66, 10325–10331. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.; Lambros, M.B.; Horlings, H.M.; Pearson, A.; Sharpe, R.; Natrajan, R.; Geyer, F.C.; van Kouwenhove, M.; Kreike, B.; Mackay, A.; et al. Integrative molecular profiling of triple negative breast cancers identifies amplicon drivers and potential thera-peutic targets. Oncogene 2010, 29, 2013–2023. [Google Scholar] [CrossRef] [Green Version]
- Yaswen, P.; Stampfer, M.R. Molecular changes accompanying senescence and immortalization of cultured human mammary epithelial cells. Int. J. Biochem. Cell Biol. 2002, 34, 1382–1394. [Google Scholar] [CrossRef]
- Dimri, G.; Band, H.; Band, V. Mammary epithelial cell transformation: Insights from cell culture and mouse models. Breast Cancer Res. 2005, 7, 171–179. [Google Scholar] [CrossRef] [Green Version]
- Soule, H.D.; Maloney, T.M.; Wolman, S.R.; Peterson, W.D.; Brenz, R.; McGrath, C.M.; Russo, J.; Pauley, R.J.; Jones, R.F.; Brooks, S.C. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990, 50, 6075–6086. [Google Scholar]
- Kadota, M.; Yang, H.H.; Gomez, B.; Sato, M.; Clifford, R.J.; Meerzaman, D.; Dunn, B.K.; Wakefield, L.M.; Lee, M.P. Delineating genetic alterations for tumor progression in the MCF10A series of breast cancer cell lines. PLoS ONE 2010, 5, e9201. [Google Scholar] [CrossRef]
- Dawson, P.J.; Wolman, S.R.; Tait, L.; Heppner, G.H.; Miller, F.R. MCF10AT: A model for the evolution of cancer from proliferative breast disease. Am. J. Pathol. 1996, 148, 313. [Google Scholar]
- Geyer, F.C.; Li, A.; Papanastasiou, A.D.; Smith, A.; Selenica, P.; Burke, K.A.; Edelweiss, M.; Wen, H.C.; Piscuoglio, S.; Schultheis, A.M.; et al. Recurrent hotspot mutations in HRAS Q61 and PI3K-AKT pathway genes as drivers of breast adenomyoepi-theliomas. Nat. Commun. 2018, 9, 1816. [Google Scholar] [CrossRef] [Green Version]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of cellular senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.; Zhang, J.; Crawford, Y.G.; Gauthier, M.L.; Fordyce, C.A.; McDermott, K.M.; Sigaroudinia, M.; Kozakiewicz, K.; Tlsty, T.D. Genetic and epigenetic changes in mammary epithelial cells identify a subpopulation of cells involved in early carcinogenesis. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 317–327. [Google Scholar] [CrossRef] [Green Version]
- Georgakopoulou, E.; Evangelou, K.; Havaki, S.; Townsend, P.; Kanavaros, P.; Gorgoulis, V.G. Apoptosis or senescence? Which exit route do epithelial cells and fibroblasts preferentially follow? Mech. Ageing Dev. 2016, 156, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Gil, J.; Efeyan, A.; Guerra, C.; Schuhmacher, A.J.; Barradas, M.; Benguría, A.; Zaballos, A.; Flores, J.M.; Barbacid, M.; et al. Senescence in premalignant tumours. Nature 2005, 436, 642. [Google Scholar] [CrossRef] [PubMed]
- Elmore, L.W.; Rehder, C.W.; Di, X.; McChesney, P.A.; Jackson-Cook, C.K.; Gewirtz, D.A.; Holt, S.E. Adriamycin-induced senescence in breast tumor cells involves functional p53 and telomere dysfunction. J. Biol. Chem. 2002, 277, 35509–35515. [Google Scholar] [CrossRef] [Green Version]
- Zacarias-Fluck, M.F.; Morancho, B.; Vicario, R.; Luque Garcia, A.; Escorihuela, M.; Villanueva, J.; Rubio, I.T.; Arribas, J. Effect of cellular senescence on the growth of HER2-positive breast cancers. J. Natl. Cancer Inst. 2015, 107, djv020. [Google Scholar] [CrossRef] [Green Version]
- McDermott, M.S.; Conlon, N.; Browne, B.C.; Szabo, A.; Synnott, N.C.; O’Brien, N.A.; Duffy, M.J.; Crown, J.; O’Donovan, N. HER2-targeted tyrosine kinase inhibitors cause therapy-induced-senescence in breast cancer cells. Cancers 2019, 11, 197. [Google Scholar] [CrossRef] [Green Version]
- Basu, A.; Pal, D.; Blaydes, R. Differential effects of protein kinase C-eta on apoptosis versus senescence. Cell. Signal. 2019, 55, 1–7. [Google Scholar] [CrossRef]
- te Poele, R.H.; Okorokov, A.L.; Jardine, L.; Cummings, J.; Joel, S.P. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res. 2002, 62, 1876–1883. [Google Scholar]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Georgakopoulou, E.A.; Tsimaratou, K.; Evangelou, K.; Fernandez, M.P.; Zoumpourlis, V.; Trougakos, I.P.; Kletsas, D.; Bartek, J.; Serrano, M.; Gorgoulis, V.G. Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues. Aging 2013, 5, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evangelou, K.; Gorgoulis, V.G. Sudan Black B, the specific histochemical stain for lipofuscin: A novel method to detect senescent cells. In Oncogene-Induced Senescence; Humana Press: New York, NY, USA, 2017; pp. 111–119. [Google Scholar]
- Evangelou, K.; Lougiakis, N.; Rizou, S.V.; Kotsinas, A.; Kletsas, D.; Muñoz-Espín, D.; Kastrinakis, N.G.; Pouli, N.; Marakos, P.; Townsend, P.; et al. Robust, universal biomarker assay to detect senescent cells in biological specimens. Aging Cell 2017, 16, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Krtolica, A.; Campisi, J. Cancer and aging: A model for the cancer promoting effects of the aging stroma. Int. J. Biochem. Cell Biol. 2002, 34, 1401–1414. [Google Scholar] [CrossRef]
- Haviv, I.; Polyak, K.; Qiu, W.; Hu, M.; Campbell, I. Origin of carcinoma associated fibroblasts. Cell Cycle 2009, 8, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Ma, C.; Hsueh, E.C.; Eickhoff, C.S.; Zhang, Y.; Varvares, M.A.; Hoft, D.F.; Peng, G. Tumor-derived γδ regulatory T cells suppress innate and adaptive immunity through the induction of immunosenescence. J. Immunol. 2013, 190, 2403–2414. [Google Scholar] [CrossRef]
- Solinas, C.; Carbognin, L.; De Silva, P.; Criscitiello, C.; Lambertini, M. Tumor-infiltrating lymphocytes in breast cancer according to tumor subtype: Current state of the art. Breast 2017, 35, 142–150. [Google Scholar] [CrossRef]
- Martins, D.; Schmitt, F. Microenvironment in breast tumorigenesis: Friend or foe? Histol. Histopathol. 2019, 34, 13–24. [Google Scholar]
- Giovannelli, P.; Di Donato, M.; Galasso, G.; Di Zazzo, E.; Medici, N.; Bilancio, A.; Migliaccio, A.; Castoria, G. Breast cancer stem cells: The role of sex steroid receptors. World J. Stem Cells 2019, 11, 594. [Google Scholar] [CrossRef]
- Schmitt, C.A. Cellular senescence and cancer treatment. Biochim. Biophys. Acta Rev. Cancer 2007, 1775, 5–20. [Google Scholar] [CrossRef]
- Trost, T.M.; Lausch, E.U.; Fees, S.A.; Schmitt, S.; Enklaar, T.; Reutzel, D.; Brixel, L.R.; Schmidtke, P.; Maringer, M.; Schiffer, I.B.; et al. Premature senescence is a primary fail-safe mechanism of ERBB2-driven tumorigenesis in breast carcinoma cells. Cancer Res. 2005, 65, 840–849. [Google Scholar]
- Jackson, J.G.; Pant, V.; Li, Q.; Chang, L.L.; Quintás-Cardama, A.; Garza, D.; Tavana, O.; Yang, P.; Manshouri, T.; Li, Y.; et al. p53-mediated senescence impairs the apoptotic response to chemotherapy and clinical outcome in breast cancer. Cancer Cell 2012, 21, 793–806. [Google Scholar] [CrossRef] [Green Version]
- Khongkow, P.; Karunarathna, U.; Khongkow, M.; Gong, C.; Gomes, A.R.; Yagüe, E.; Monteiro, L.J.; Kongsema, M.; Zona, S.; Man, E.P.; et al. FOXM1 targets NBS1 to regulate DNA damage-induced senescence and epirubicin resistance. Oncogene 2014, 33, 4144–4155. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Dokmanovic, M.; Stein, W.D.; Ardecky, R.J.; Roninson, I.B. Agonist and antagonist of retinoic acid receptors cause similar changes in gene expression and induce senescence-like growth arrest in MCF-7 breast carcinoma cells. Cancer Res. 2006, 66, 8749–8761. [Google Scholar] [CrossRef] [Green Version]
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-induced senescence in cancer. J. Natl. Cancer Inst. 2010, 102, 1536–1546. [Google Scholar] [CrossRef] [Green Version]
- Hoenicke, L.; Zender, L. Immune surveillance of senescent cells—Biological significance in cancer-and non-cancer pathologies. Carcinogenesis 2012, 33, 1123–1126. [Google Scholar] [CrossRef] [Green Version]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.U.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; Van De Sluis, B.; Kirkland, J.L.; Van Deursen, J.M. Clearance of p16 Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Sun, Y.; Campisi, J.; Higano, C.; Beer, T.M.; Porter, P.; Coleman, I.; True, L.; Nelson, P.S. Treatment-induced damage to the tumor mi-croenvironment promotes prostate cancer therapy resistance through WNT16B. Nature Med. 2012, 18, 1359–1368. [Google Scholar] [CrossRef]
- Sanoff, H.K.; Deal, A.M.; Krishnamurthy, J.; Torrice, C.; Dillon, P.; Sorrentino, J.; Ibrahim, J.G.; Jolly, T.A.; Williams, G.; Carey, L.A.; et al. Effect of cytotoxic chemotherapy on markers of molecular age in patients with breast cancer. J. Natl. Cancer Inst. 2014, 106, dju057. [Google Scholar] [CrossRef] [Green Version]
- Childs, B.G.; Durik, M.; Baker, D.J.; Van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [Green Version]
- Reddy, J.P.; Li, Y. Oncogene-induced senescence and its role in tumor suppression. J. Mammary Gland. Biol. Neo-Plasia 2011, 16, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.A.; Rubelj, I.; Pereira-Smith, O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [Green Version]
- Narita, M.; Nuñez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef] [Green Version]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular senescence: Defining a path forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef]
- Myrianthopoulos, V.; Evangelou, K.; Vasileiou, P.V.; Cooks, T.; Vassilakopoulos, T.P.; Pangalis, G.A.; Kouloukoussa, M.; Kittas, C.; Georgakilas, A.G.; Gorgoulis, V.G. Senescence and senotherapeutics: A new field in cancer therapy. Pharmacol. Ther. 2019, 193, 31–49. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sirinian, C.; Peroukidis, S.; Kriegsmann, K.; Chaniotis, D.; Koutras, A.; Kriegsmann, M.; Papanastasiou, A.D. Cellular Senescence in Normal Mammary Gland and Breast Cancer. Implications for Cancer Therapy. Genes 2022, 13, 994. https://doi.org/10.3390/genes13060994
Sirinian C, Peroukidis S, Kriegsmann K, Chaniotis D, Koutras A, Kriegsmann M, Papanastasiou AD. Cellular Senescence in Normal Mammary Gland and Breast Cancer. Implications for Cancer Therapy. Genes. 2022; 13(6):994. https://doi.org/10.3390/genes13060994
Chicago/Turabian StyleSirinian, Chaido, Stavros Peroukidis, Katharina Kriegsmann, Dimitrios Chaniotis, Angelos Koutras, Mark Kriegsmann, and Anastasios D. Papanastasiou. 2022. "Cellular Senescence in Normal Mammary Gland and Breast Cancer. Implications for Cancer Therapy" Genes 13, no. 6: 994. https://doi.org/10.3390/genes13060994