
Dynamic Features of Chromosomal Instability during Culture of Induced Pluripotent Stem Cells


Abstract
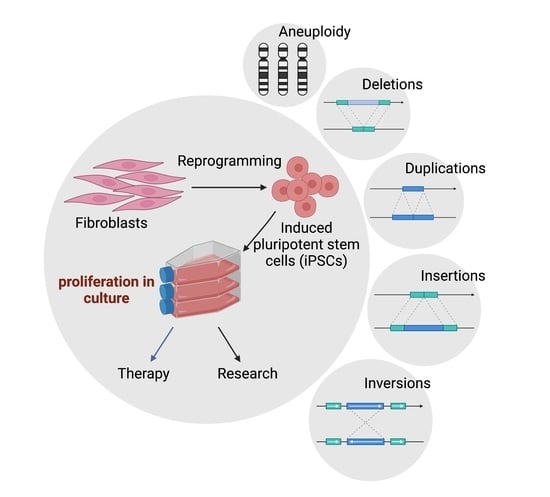
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. IPSC Growth Rate and Aneuploidy
2.2. Structural Variant Detection, Filtration and Characterization
2.3. Structural Variation Impact on Gene Function
3. Prospects and Conclusions
4. Methods
4.1. IPS Cell Lines
4.2. Cell Culture
4.3. Chromosome Spreads
4.4. Predictive Mathematical Modeling
5. Bionano Genomics Techniques
5.1. Genomic DNA Isolation
5.2. DNA Labeling
5.3. Chip Loading and Analysis
5.4. Structural Variant Analysis Software
5.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merkle, F.T.; Ghosh, S.; Kamitaki, N.; Mitchell, J.; Avior, Y.; Mello, C.; Kashin, S.; Mekhoubad, S.; Ilic, D.; Charlton, M.; et al. Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature 2017, 545, 229–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuijk, E.; Jager, M.; van der Roest, B.; Locati, M.D.; Van Hoeck, A.; Korzelius, J.; Janssen, R.; Besselink, N.; Boymans, S.; van Boxtel, R.; et al. The mutational impact of culturing human pluripotent and adult stem cells. Nat. Commun. 2020, 11, 2493. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, J.; Barbaric, I.; Andrews, P.W. Acquired genetic changes in human pluripotent stem cells: Origins and consequences. Nat. Rev. Mol. Cell Biol. 2020, 21, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Steichen, C.; Hannoun, Z.; Luce, E.; Hauet, T.; Dubart-Kupperschmitt, A. Genomic integrity of human induced pluripotent stem cells: Reprogramming, differentiation and applications. World J. Stem Cells 2019, 11, 729–747. [Google Scholar] [CrossRef]
- Catalina, P.; Montes, R.; Ligero, G.; Sanchez, L.; de la Cueva, T.; Bueno, C.; Leone, P.E.; Menendez, P. Human ESCs predisposition to karyotypic instability: Is a matter of culture adaptation or differential vulnerability among hESC lines due to inherent properties? Mol. Cancer 2008, 7, 76. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Kaplan, A.; Yuan, B.; Hanna, J.H.; Lupski, J.R.; Reiner, O. Passage number is a major contributor to genomic structural variations in mouse iPSCs. Stem Cells 2014, 32, 2657–2667. [Google Scholar] [CrossRef] [Green Version]
- Martins-Taylor, K.; Nisler, B.S.; Taapken, S.M.; Compton, T.; Crandall, L.; Montgomery, K.D.; Lalande, M.; Xu, R.H. Recurrent copy number variations in human induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 488–491. [Google Scholar] [CrossRef]
- Mayshar, Y.; Ben-David, U.; Lavon, N.; Biancotti, J.C.; Yakir, B.; Clark, A.T.; Plath, K.; Lowry, W.E.; Benvenisty, N. Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell 2010, 7, 521–531. [Google Scholar] [CrossRef] [Green Version]
- Rebuzzini, P.; Zuccotti, M.; Redi, C.A.; Garagna, S. Achilles’ heel of pluripotent stem cells: Genetic, genomic and epigenetic variations during prolonged culture. Cell Mol. Life Sci. 2016, 73, 2453–2466. [Google Scholar] [CrossRef]
- Simara, P.; Tesarova, L.; Rehakova, D.; Matula, P.; Stejskal, S.; Hampl, A.; Koutna, I. DNA double-strand breaks in human induced pluripotent stem cell reprogramming and long-term in vitro culturing. Stem Cell Res. Ther. 2017, 8, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taapken, S.M.; Nisler, B.S.; Newton, M.A.; Sampsell-Barron, T.L.; Leonhard, K.A.; McIntire, E.M.; Montgomery, K.D. Karotypic abnormalities in human induced pluripotent stem cells and embryonic stem cells. Nat. Biotechnol. 2011, 29, 313–314. [Google Scholar] [CrossRef] [PubMed]
- Vallabhaneni, H.; Lynch, P.J.; Chen, G.; Park, K.; Liu, Y.; Goehe, R.; Mallon, B.S.; Boehm, M.; Hursh, D.A. High Basal Levels of gammaH2AX in Human Induced Pluripotent Stem Cells Are Linked to Replication-Associated DNA Damage and Repair. Stem Cells 2018, 36, 1501–1513. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.E.; Harrison, N.J.; Maltby, E.; Smith, K.; Moore, H.D.; Shaw, P.J.; Heath, P.R.; Holden, H.; Andrews, P.W. Adaptation to culture of human embryonic stem cells and oncogenesis in vivo. Nat. Biotechnol. 2007, 25, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Amariglio, N.; Hirshberg, A.; Scheithauer, B.W.; Cohen, Y.; Loewenthal, R.; Trakhtenbrot, L.; Paz, N.; Koren-Michowitz, M.; Waldman, D.; Leider-Trejo, L.; et al. Donor-derived brain tumor following neural stem cell transplantation in an ataxia telangiectasia patient. PLoS Med. 2009, 6, e1000029. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.W.; Ben-David, U.; Benvenisty, N.; Coffey, P.; Eggan, K.; Knowles, B.B.; Nagy, A.; Pera, M.; Reubinoff, B.; Rugg-Gunn, P.J.; et al. Assessing the Safety of Human Pluripotent Stem Cells and Their Derivatives for Clinical Applications. Stem Cell Rep. 2017, 9, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Ben-David, U.; Benvenisty, N. The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat. Rev. Cancer 2011, 11, 268–277. [Google Scholar] [CrossRef]
- Werbowetski-Ogilvie, T.E.; Schnerch, A.; Rampalli, S.; Mills, C.E.; Lee, J.B.; Hong, S.H.; Levadoux-Martin, M.; Bhatia, M. Evidence for the transmission of neoplastic properties from transformed to normal human stem cells. Oncogene 2011, 30, 4632–4644. [Google Scholar] [CrossRef] [Green Version]
- Ben-David, U.; Arad, G.; Weissbein, U.; Mandefro, B.; Maimon, A.; Golan-Lev, T.; Narwani, K.; Clark, A.T.; Andrews, P.W.; Benvenisty, N.; et al. Aneuploidy induces profound changes in gene expression, proliferation and tumorigenicity of human pluripotent stem cells. Nat. Commun. 2014, 5, 4825. [Google Scholar] [CrossRef]
- Garcia-Martinez, J.; Bakker, B.; Schukken, K.M.; Simon, J.E.; Foijer, F. Aneuploidy in stem cells. World J. Stem Cells 2016, 8, 216–222. [Google Scholar] [CrossRef]
- Keller, A.; Spits, C. The Impact of Acquired Genetic Abnormalities on the Clinical Translation of Human Pluripotent Stem Cells. Cells 2021, 10, 3246. [Google Scholar] [CrossRef] [PubMed]
- International Stem Cell, I.; Amps, K.; Andrews, P.W.; Anyfantis, G.; Armstrong, L.; Avery, S.; Baharvand, H.; Baker, J.; Baker, D.; Munoz, M.B.; et al. Screening ethnically diverse human embryonic stem cells identifies a chromosome 20 minimal amplicon conferring growth advantage. Nat. Biotechnol. 2011, 29, 1132–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Draper, J.S.; Smith, K.; Gokhale, P.; Moore, H.D.; Maltby, E.; Johnson, J.; Meisner, L.; Zwaka, T.P.; Thomson, J.A.; Andrews, P.W. Recurrent gain of chromosomes 17q and 12 in cultured human embryonic stem cells. Nat. Biotechnol. 2004, 22, 53–54. [Google Scholar] [CrossRef]
- Mills, R.E.; Walter, K.; Stewart, C.; Handsaker, R.E.; Chen, K.; Alkan, C.; Abyzov, A.; Yoon, S.C.; Ye, K.; Cheetham, R.K.; et al. Mapping copy number variation by population-scale genome sequencing. Nature 2011, 470, 59–65. [Google Scholar] [CrossRef]
- Pang, A.W.; MacDonald, J.R.; Pinto, D.; Wei, J.; Rafiq, M.A.; Conrad, D.F.; Park, H.; Hurles, M.E.; Lee, C.; Venter, J.C.; et al. Towards a comprehensive structural variation map of an individual human genome. Genome Biol. 2010, 11, R52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Vries, B.B.; Pfundt, R.; Leisink, M.; Koolen, D.A.; Vissers, L.E.; Janssen, I.M.; Reijmersdal, S.; Nillesen, W.M.; Huys, E.H.; Leeuw, N.; et al. Diagnostic genome profiling in mental retardation. Am. J. Hum. Genet. 2005, 77, 606–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, C.R.; Noor, A.; Vincent, J.B.; Lionel, A.C.; Feuk, L.; Skaug, J.; Shago, M.; Moessner, R.; Pinto, D.; Ren, Y.; et al. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 2008, 82, 477–488. [Google Scholar] [CrossRef] [Green Version]
- Lupski, J.R. Genomic disorders: Structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998, 14, 417–422. [Google Scholar] [CrossRef]
- Weischenfeldt, J.; Symmons, O.; Spitz, F.; Korbel, J.O. Phenotypic impact of genomic structural variation: Insights from and for human disease. Nat. Rev. Genet. 2013, 14, 125–138. [Google Scholar] [CrossRef]
- Muller, V.; Westerlund, F. Optical DNA mapping in nanofluidic devices: Principles and applications. Lab Chip 2017, 17, 579–590. [Google Scholar] [CrossRef]
- Shakiba, N.; Zandstra, P.W. Engineering cell fitness: Lessons for regenerative medicine. Curr. Opin. Biotechnol. 2017, 47, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Bowling, S.; Lawlor, K.; Rodriguez, T.A. Cell competition: The winners and losers of fitness selection. Development 2019, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morata, G.; Ripoll, P. Minutes: Mutants of drosophila autonomously affecting cell division rate. Dev. Biol. 1975, 42, 211–221. [Google Scholar] [CrossRef]
- Price, C.J.; Stavish, D.; Gokhale, P.J.; Stevenson, B.A.; Sargeant, S.; Lacey, J.; Rodriguez, T.A.; Barbaric, I. Genetically variant human pluripotent stem cells selectively eliminate wild-type counterparts through YAP-mediated cell competition. Dev. Cell 2021, 56, 2455–2470.e2410. [Google Scholar] [CrossRef] [PubMed]
- Kreitzer, F.R.; Salomonis, N.; Sheehan, A.; Huang, M.; Park, J.S.; Spindler, M.J.; Lizarraga, P.; Weiss, W.A.; So, P.L.; Conklin, B.R. A robust method to derive functional neural crest cells from human pluripotent stem cells. Am. J. Stem Cells 2013, 2, 119–131. [Google Scholar]
- Kilpinen, H.; Goncalves, A.; Leha, A.; Afzal, V.; Alasoo, K.; Ashford, S.; Bala, S.; Bensaddek, D.; Casale, F.P.; Culley, O.J.; et al. Common genetic variation drives molecular heterogeneity in human iPSCs. Nature 2017, 546, 370–375. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.; Hirst, A.J.; Gokhale, P.J.; Juarez, M.A.; Williams, S.; Wheeler, M.; Bean, K.; Allison, T.F.; Moore, H.D.; Andrews, P.W.; et al. Detecting Genetic Mosaicism in Cultures of Human Pluripotent Stem Cells. Stem Cell Rep. 2016, 7, 998–1012. [Google Scholar] [CrossRef] [Green Version]
- Laurent, L.C.; Ulitsky, I.; Slavin, I.; Tran, H.; Schork, A.; Morey, R.; Lynch, C.; Harness, J.V.; Lee, S.; Barrero, M.J.; et al. Dynamic changes in the copy number of pluripotency and cell proliferation genes in human ESCs and iPSCs during reprogramming and time in culture. Cell Stem Cell 2011, 8, 106–118. [Google Scholar] [CrossRef] [Green Version]
- Genomes Project, C.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Tsai, H.J.; Gordon, M.R.; Li, R. Cellular Stress Associated with Aneuploidy. Dev. Cell 2018, 44, 420–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, A.J.; Brennan, C.M.; Xu, A.E.; Solis, E.J.; Terhorst, A.; Denic, V.; Amon, A. Cell adaptation to aneuploidy by the environmental stress response dampens induction of the cytosolic unfolded-protein response. Mol. Biol. Cell 2021, 32, 1557–1564. [Google Scholar] [CrossRef] [PubMed]
- Pavelka, N.; Rancati, G.; Zhu, J.; Bradford, W.D.; Saraf, A.; Florens, L.; Sanderson, B.W.; Hattem, G.L.; Li, R. Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature 2010, 468, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Torres, E.M.; Sokolsky, T.; Tucker, C.M.; Chan, L.Y.; Boselli, M.; Dunham, M.J.; Amon, A. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 2007, 317, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.L.; Brand, H.; Karczewski, K.J.; Zhao, X.; Alfoldi, J.; Francioli, L.C.; Khera, A.V.; Lowther, C.; Gauthier, L.D.; Wang, H.; et al. A structural variation reference for medical and population genetics. Nature 2020, 581, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, L.G.; Lupski, J.R. Molecular mechanisms for constitutional chromosomal rearrangements in humans. Annu. Rev. Genet. 2000, 34, 297–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Li, H.; Wang, Y.; Wang, J.; Zheng, Q.; Hua, J.; Yang, J.; Pan, L.; Lu, F.; Qu, J.; et al. DAPL1, a susceptibility locus for age-related macular degeneration, acts as a novel suppressor of cell proliferation in the retinal pigment epithelium. Hum. Mol. Genet. 2017, 26, 1612–1621. [Google Scholar] [CrossRef]
- Grassmann, F.; Friedrich, U.; Fauser, S.; Schick, T.; Milenkovic, A.; Schulz, H.L.; von Strachwitz, C.N.; Bettecken, T.; Lichtner, P.; Meitinger, T.; et al. A Candidate Gene Association Study Identifies DAPL1 as a Female-Specific Susceptibility Locus for Age-Related Macular Degeneration (AMD). Neuromolecular Med. 2015, 17, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 2009, 4, 44–57. [Google Scholar] [CrossRef]
- McKee, C.; Chaudhry, G.R. Advances and challenges in stem cell culture. Colloids Surf. B Biointerfaces 2017, 159, 62–77. [Google Scholar] [CrossRef]
- Watanabe, K.; Ueno, M.; Kamiya, D.; Nishiyama, A.; Matsumura, M.; Wataya, T.; Takahashi, J.B.; Nishikawa, S.; Nishikawa, S.; Muguruma, K.; et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat. Biotechnol. 2007, 25, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Holm, F.; Nikdin, H.; Kjartansdottir, K.R.; Gaudenzi, G.; Fried, K.; Aspenstrom, P.; Hermanson, O.; Bergstrom-Tengzelius, R. Passaging techniques and ROCK inhibitor exert reversible effects on morphology and pluripotency marker gene expression of human embryonic stem cell lines. Stem Cells Dev. 2013, 22, 1883–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Q.; Ramirez, J.M.; Becker, F.; Pantesco, V.; Lavabre-Bertrand, T.; Hovatta, O.; Lemaitre, J.M.; Pellestor, F.; De Vos, J. Temporal analysis of genome alterations induced by single-cell passaging in human embryonic stem cells. Stem Cells Dev. 2015, 24, 653–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garitaonandia, I.; Amir, H.; Boscolo, F.S.; Wambua, G.K.; Schultheisz, H.L.; Sabatini, K.; Morey, R.; Waltz, S.; Wang, Y.C.; Tran, H.; et al. Increased risk of genetic and epigenetic instability in human embryonic stem cells associated with specific culture conditions. PLoS ONE 2015, 10, e0118307. [Google Scholar] [CrossRef]
- Maitra, A.; Arking, D.E.; Shivapurkar, N.; Ikeda, M.; Stastny, V.; Kassauei, K.; Sui, G.; Cutler, D.J.; Liu, Y.; Brimble, S.N.; et al. Genomic alterations in cultured human embryonic stem cells. Nat. Genet. 2005, 37, 1099–1103. [Google Scholar] [CrossRef]
- Tosca, L.; Feraud, O.; Magniez, A.; Bas, C.; Griscelli, F.; Bennaceur-Griscelli, A.; Tachdjian, G. Genomic instability of human embryonic stem cell lines using different passaging culture methods. Mol. Cytogenet. 2015, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Mitalipova, M.M.; Rao, R.R.; Hoyer, D.M.; Johnson, J.A.; Meisner, L.F.; Jones, K.L.; Dalton, S.; Stice, S.L. Preserving the genetic integrity of human embryonic stem cells. Nat. Biotechnol. 2005, 23, 19–20. [Google Scholar] [CrossRef]
- Poetsch, M.S.; Strano, A.; Guan, K. Human induced pluripotent stem cells: From cell origin, genomic stability and epigenetic memory to translational medicine. Stem Cells 2022, 40, 546–555. [Google Scholar] [CrossRef]
- Akutsu, S.N.; Miyamoto, T.; Oba, D.; Tomioka, K.; Ochiai, H.; Ohashi, H.; Matsuura, S. iPSC reprogramming-mediated aneuploidy correction in autosomal trisomy syndromes. PLoS ONE 2022, 17, e0264965. [Google Scholar] [CrossRef]
- Inoue, M.; Kajiwara, K.; Yamaguchi, A.; Kiyono, T.; Samura, O.; Akutsu, H.; Sago, H.; Okamoto, A.; Umezawa, A. Autonomous trisomic rescue of Down syndrome cells. Lab. Investig. 2019, 99, 885–897. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
DuBose, C.O.; Daum, J.R.; Sansam, C.L.; Gorbsky, G.J. Dynamic Features of Chromosomal Instability during Culture of Induced Pluripotent Stem Cells. Genes 2022, 13, 1157. https://doi.org/10.3390/genes13071157
DuBose CO, Daum JR, Sansam CL, Gorbsky GJ. Dynamic Features of Chromosomal Instability during Culture of Induced Pluripotent Stem Cells. Genes. 2022; 13(7):1157. https://doi.org/10.3390/genes13071157
Chicago/Turabian StyleDuBose, Casey O., John R. Daum, Christopher L. Sansam, and Gary J. Gorbsky. 2022. "Dynamic Features of Chromosomal Instability during Culture of Induced Pluripotent Stem Cells" Genes 13, no. 7: 1157. https://doi.org/10.3390/genes13071157