Methionine Adenosyltransferase I/III Deficiency Detected by Newborn Screening

 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
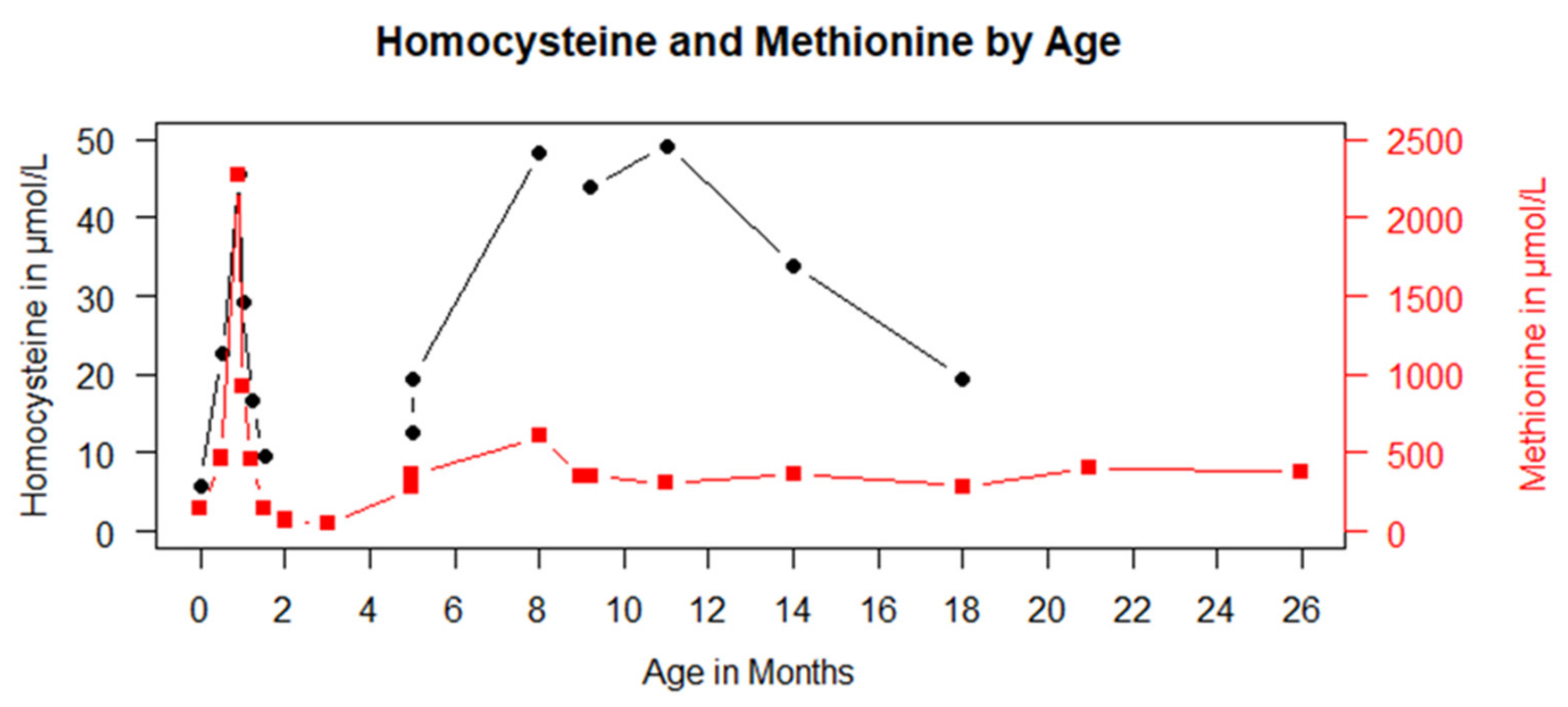
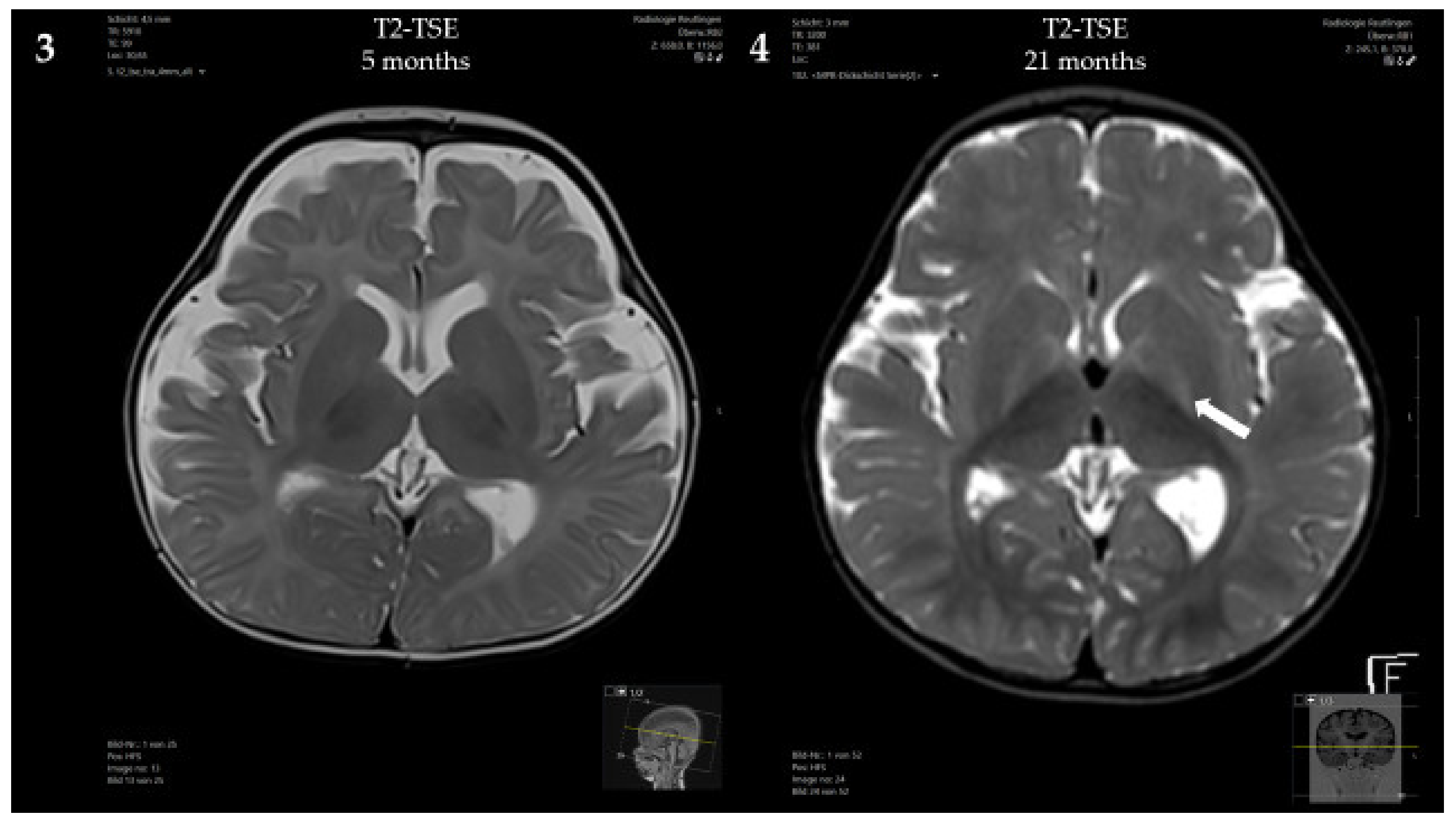
3.1. Case Presentation
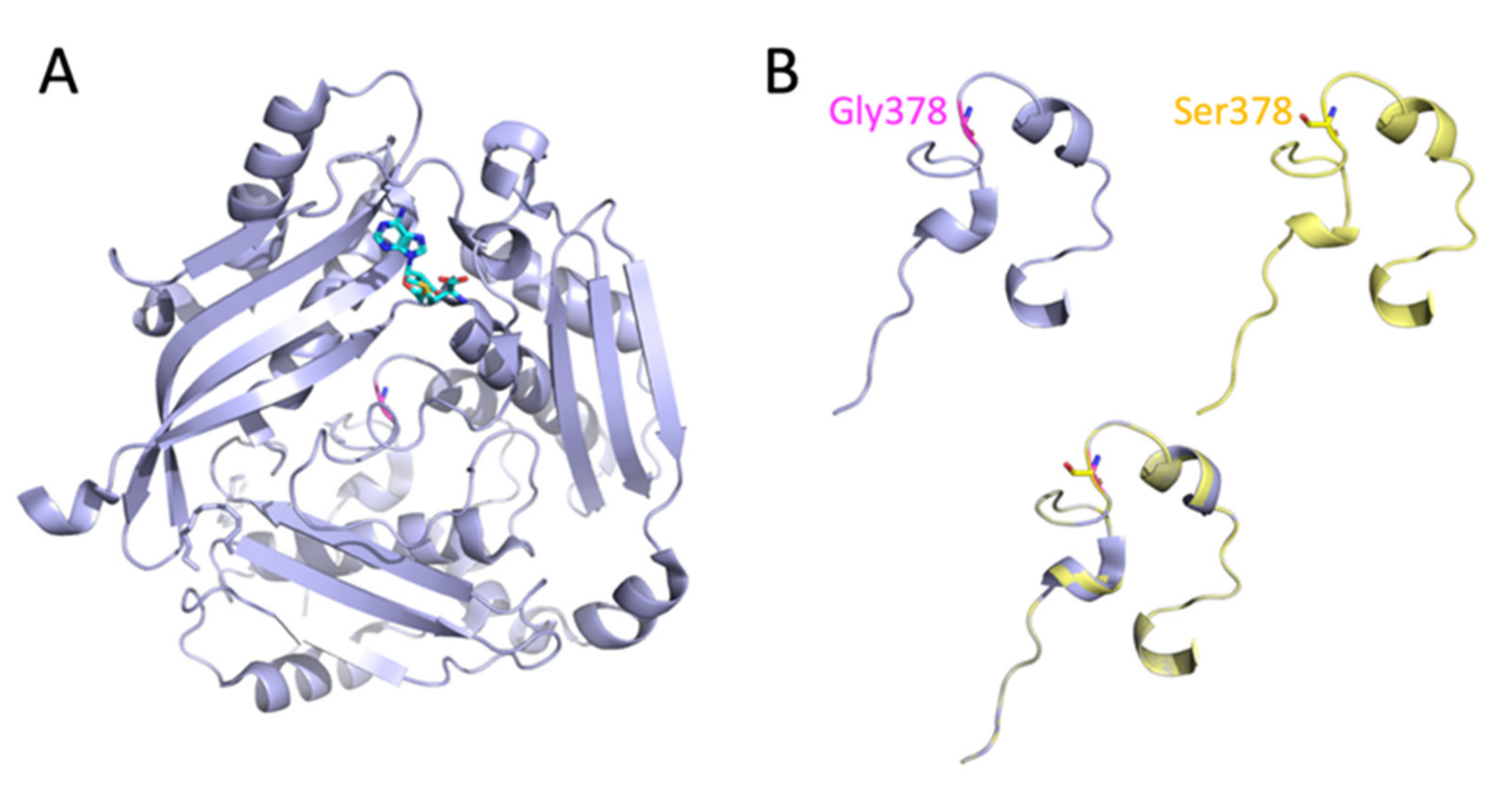
3.2. Analysis of the MAT1A Protein Structure
3.3. Predicted Effect of the Homozygous c.1132G>A; p.Gly378Ser Mutation on the Enzymatic Activity of MAT I/III
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, Y.M.; Kim, J.H.; Choi, J.H.; Kim, G.H.; Kim, J.M.; Kang, M.; Choi, I.H.; Cheon, C.K.; Sohn, Y.B.; Maccarana, M.; et al. Determination of Autosomal Dominant or Recessive Methionine Adenosyltransferase I/III Deficiencies Based on Clinical and Molecular Studies. Mol. Med. 2016, 22, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Mudd, S.H. Hypermethioninemias of genetic and non-genetic origin: A review. Am. J. Med. Genet. C Semin. Med. Genet. 2011, 157, 3–32. [Google Scholar] [CrossRef] [PubMed]
- Lauinger, L.; Kaiser, P. Sensing and Signaling of Methionine Metabolism. Metabolites 2021, 11, 83. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.-H.; Abdenur, J.E.; Baronio, F.; Bannick, A.A.; Corrales, F.; Couce, M.; Donner, M.G.; Ficicioglu, C.; Freehauf, C.; Frithiof, D.; et al. Mudd’s disease (MAT I/III deficiency): A survey of data for MAT1A homozygotes and compound heterozygotes. Orphanet J. Rare Dis. 2015, 10, 99. [Google Scholar] [CrossRef] [Green Version]
- Nashabat, M.; Al-Khenaizan, S.; Alfadhel, M. Methionine adenosyltransferase I/III deficiency: Beyond the central nervous system manifestations. Ther. Clin. Risk Manag. 2018, 14, 225–229. [Google Scholar] [CrossRef] [Green Version]
- Surtees, R.; Leonard, J.; Austin, S. Association of demyelination with deficiency of cerebrospinal-fluid S-adenosylmethionine in inborn errors of methyl-transfer pathway. Lancet 1991, 338, 1550–1554. [Google Scholar] [CrossRef]
- Couce, M.L.; Bóveda, M.D.; Castiñeiras, D.E.; Corrales, F.J.; Mora, M.I.; Fraga, J.M.; Mudd, S.H. Hypermethioninaemia due to methionine adenosyltransferase I/III (MAT I/III) deficiency: Diagnosis in an expanded neonatal screening programme. J. Inherit. Metab. Dis. 2008, 31 (Suppl. S2), S233–S239. [Google Scholar] [CrossRef] [Green Version]
- Couce, M.L.; Bóveda, M.D.; García-Jimémez, C.; Balmaseda, E.; Vives, I.; Castiñeiras, D.E.; Fernández-Marmiesse, A.; Fraga, J.M.; Mudd, S.H.; Corrales, F.J. Clinical and metabolic findings in patients with methionine adenosyltransferase I/III deficiency detected by newborn screening. Mol. Genet. Metab. 2013, 110, 218–221. [Google Scholar] [CrossRef]
- Martins, E.; Marcão, A.; Bandeira, A.; Fonseca, H.; Nogueira, C.; Vilarinho, L. Methionine Adenosyltransferase I/III Deficiency in Portugal: High Frequency of a Dominantly Inherited Form in a Small Area of Douro High Lands. JIMD Rep. 2012, 6, 107–112. [Google Scholar]
- Furujo, M.; Kinoshita, M.; Nagao, M.; Kubo, T. Methionine adenosyltransferase I/III deficiency: Neurological manifestations and relevance of S-adenosylmethionine. Mol. Genet. Metab. 2012, 107, 253–256. [Google Scholar] [CrossRef]
- Barić, I.; Staufner, C.; Augoustides-Savvopoulou, P.; Chien, Y.-H.; Dobbelaere, D.; Grünert, S.C.; Opladen, T.; Ramadža, D.P.; Rakić, B.; Wedell, A.; et al. Consensus recommendations for the diagnosis, treatment and follow-up of inherited methylation disorders. J. Inherit. Metab. Dis. 2017, 40, 5–20. [Google Scholar] [CrossRef] [Green Version]
- Shafqat, N.; Muniz, J.R.C.; Pilka, E.S.; Papagrigoriou, E.; von Delft, F.; Oppermann, U.; Yue, W.W. Insight into S-adenosylmethionine biosynthesis from the crystal structures of the human methionine adenosyltransferase catalytic and regulatory subunits. Biochem. J. 2013, 452, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Markham, G.D.; Pajares, M.A. Structure-function relationships in methionine adenosyltransferases. Cell. Mol. Life Sci. 2009, 66, 636–648. [Google Scholar] [CrossRef] [Green Version]
- Houry, W.A.; Frishman, D.; Eckerskorn, C.; Lottspeich, F.; Hartl, F.U. Identification of in vivo substrates of the chaperonin GroEL. Nature 1999, 402, 147–154. [Google Scholar] [CrossRef]
- Chamberlin, M.E.; Ubagai, T.; Mudd, S.H.; Wilson, W.G.; Leonard, J.V.; Chou, J.Y. Demyelination of the brain is associated with methionine adenosyltransferase I/III deficiency. J. Clin. Investig. 1996, 98, 1021–1027. [Google Scholar] [CrossRef]
- Braverman, N.E.; Mudd, S.H.; Barker, P.B.; Pomper, M.G. Characteristic MR imaging changes in severe hypermethioninemic states. AJNR Am. J. Neuroradiol. 2005, 26, 2705–2706. [Google Scholar]
- Tada, H.; Takanashi, J.; Barkovich, A.J.; Yamamoto, S.; Kohno, Y. Reversible white matter lesion in methionine adenosyltransferase I/III deficiency. AJNR Am. J. Neuroradiol. 2004, 25, 1843–1845. [Google Scholar]
- Mudd, S.H.; Braverman, N.; Pomper, M.; Tezcan, K.; Kronick, J.; Jayakar, P.; Garganta, C.; Ampola, M.G.; Levy, H.L.; McCandless, S.E.; et al. Infantile hypermethioninemia and hyperhomocysteinemia due to high methionine intake: A diagnostic trap. Mol. Genet. Metab. 2003, 79, 6–16. [Google Scholar] [CrossRef]
- Young, S.N.; Shalchi, M. The effect of methionine and S-adenosylmethionine on S-adenosylmethionine levels in the rat brain. J. Psychiatry Neurosci. 2005, 30, 44–48. [Google Scholar]
- Chamberlin, M.E.; Ubagai, T.; Mudd, S.H.; Thomas, J.; Pao, V.Y.; Nguyen, T.K.; Levy, H.L.; Greene, C.; Freehauf, C.; Chou, J.Y. Methionine Adenosyltransferase I/III Deficiency: Novel Mutations and Clinical Variations. Am. J. Hum. Genet. 2000, 66, 347–355. [Google Scholar] [CrossRef] [Green Version]
- Inoue, K.; Khajavi, M.; Ohyama, T.; Hirabayashi, S.-I.; Wilson, J.H.; Reggin, J.D.; Mancias, P.; Butler, I.J.; Wilkinson, M.F.; Wegner, M.; et al. Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat. Genet. 2004, 36, 361–369. [Google Scholar] [CrossRef]
- Kim, S.Z.; Santamaria, E.; Jeong, T.E.; Levy, H.L.; Mato, J.M.; Corrales, F.J.; Mudd, S.H. Methionine adenosyltransferase I/III deficiency: Two Korean compound heterozygous siblings with a novel mutation. J. Inherit. Metab. Dis. 2003, 25, 661–671. [Google Scholar] [CrossRef]
- Cohen, B.M.; Renshaw, P.F.; Stoll, A.L.; Wurtman, R.J.; Yurgelun-Todd, D.; Babb, S.M. Decreased brain choline uptake in older adults. An in vivo proton magnetic resonance spectroscopy study. JAMA 1995, 274, 902–907. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hübner, V.; Hannibal, L.; Janzen, N.; Grünert, S.C.; Freisinger, P. Methionine Adenosyltransferase I/III Deficiency Detected by Newborn Screening. Genes 2022, 13, 1163. https://doi.org/10.3390/genes13071163
Hübner V, Hannibal L, Janzen N, Grünert SC, Freisinger P. Methionine Adenosyltransferase I/III Deficiency Detected by Newborn Screening. Genes. 2022; 13(7):1163. https://doi.org/10.3390/genes13071163
Chicago/Turabian StyleHübner, Vanessa, Luciana Hannibal, Nils Janzen, Sarah Catharina Grünert, and Peter Freisinger. 2022. "Methionine Adenosyltransferase I/III Deficiency Detected by Newborn Screening" Genes 13, no. 7: 1163. https://doi.org/10.3390/genes13071163