
Clinical Features and Genetic Findings of Autosomal Recessive Bestrophinopathy

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Subjects
2.2. Data Collection
2.3. Ophthalmologic Image Interpretation
2.4. Genetic Analysis
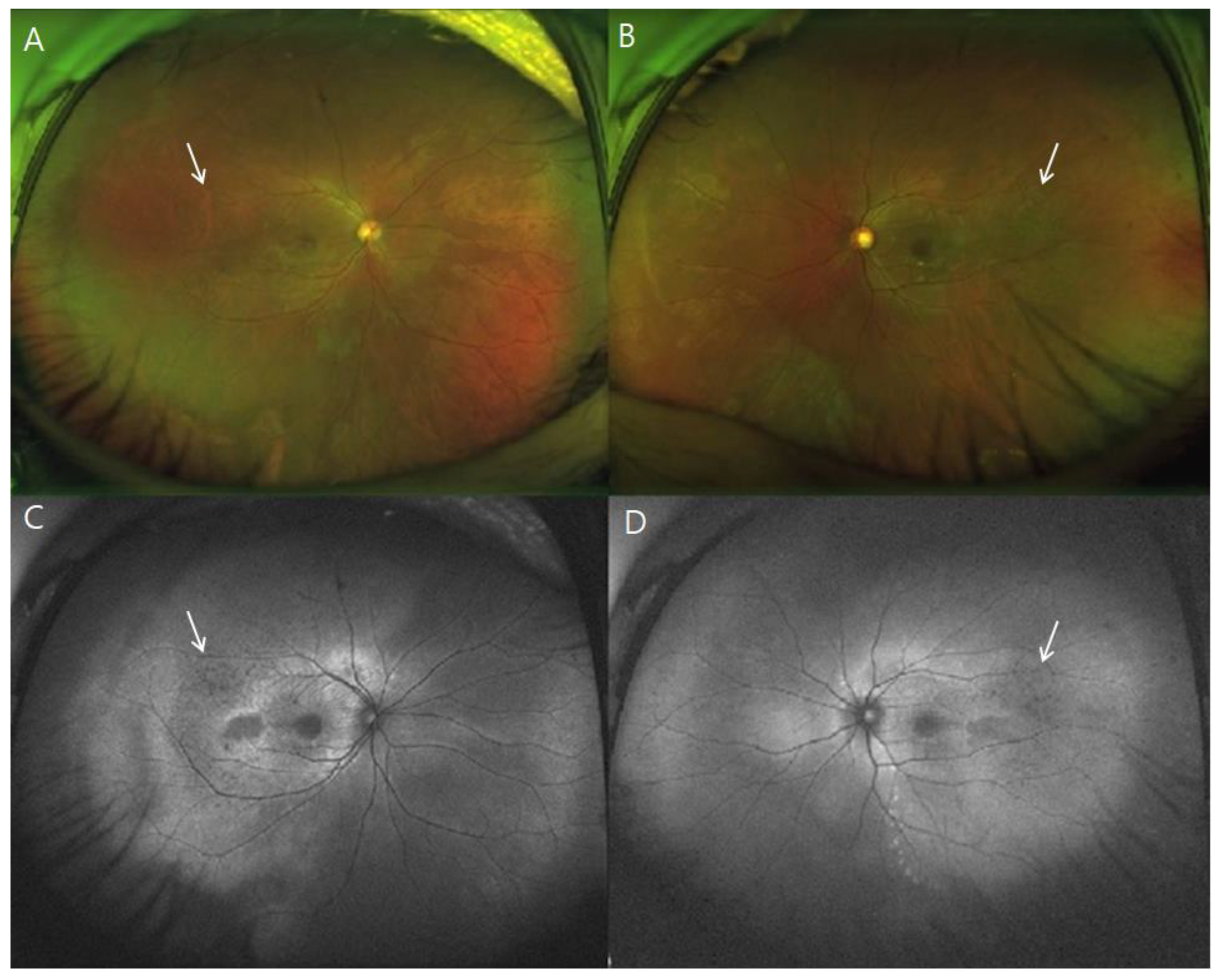
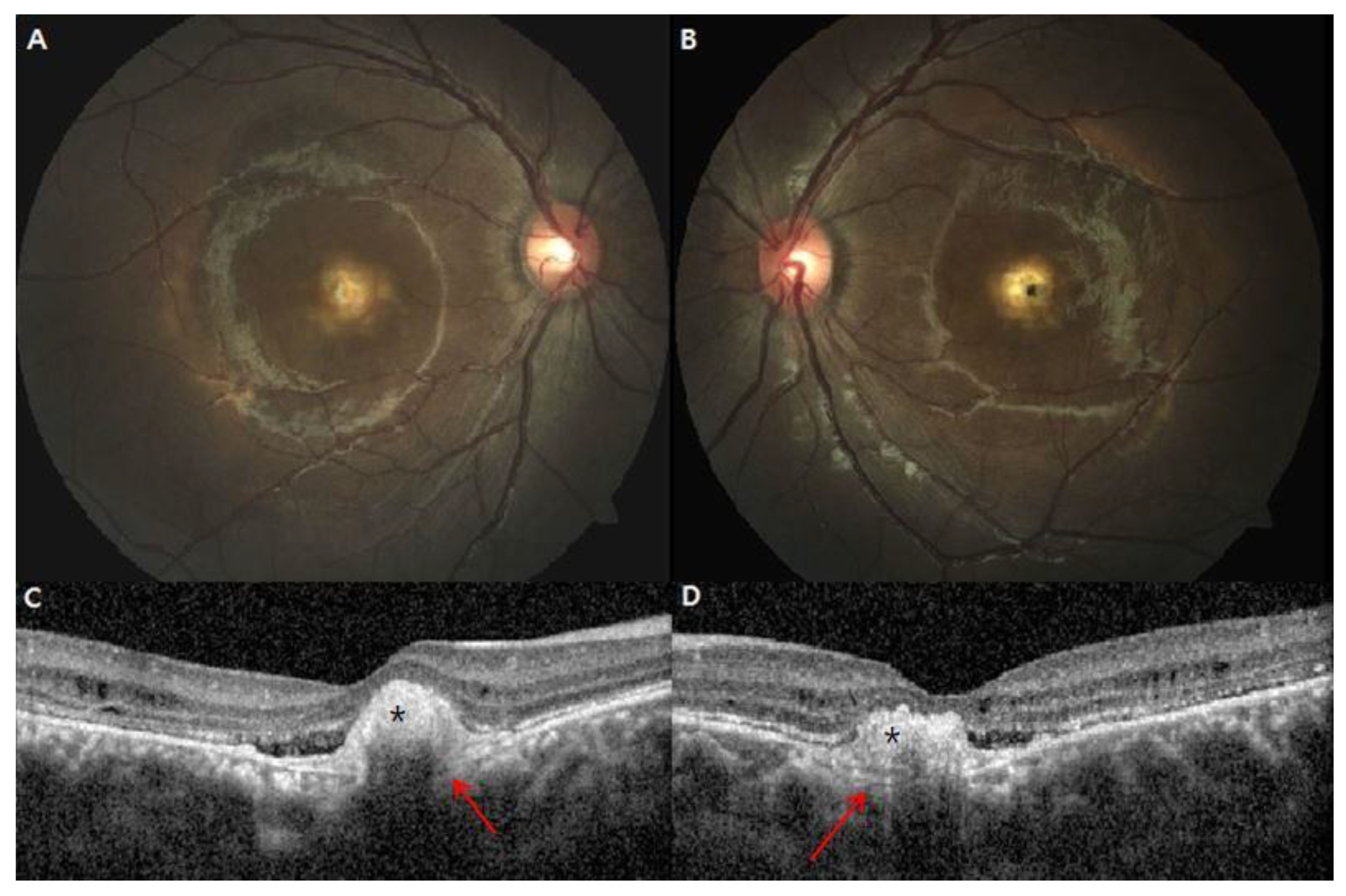
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, J.H.; Oh, J.O.; Lee, C.S. Induced Pluripotent Stem Cell Modeling of Best Disease and Autosomal Recessive Bestrophinopathy. Yonsei Med. J. 2020, 61, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Forsman, K.; Graff, C.; Nordström, S.; Johansson, K.; Westermark, E.; Lundgren, E.; Gustavson, K.H.; Wadelius, C.; Holmgren, G. The gene for Best’s macular dystrophy is located at 11q13 in a Swedish family. Clin. Genet. 1992, 42, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.M.; Nichols, B.E.; Streb, L.M.; Kimura, A.E.; Sheffield, V.C. Genetic linkage of vitelliform macular degeneration (Best’s disease) to chromosome 11q13. Nat. Genet. 1992, 1, 246–250. [Google Scholar] [CrossRef]
- Marmorstein, A.D.; Marmorstein, L.Y.; Rayborn, M.; Wang, X.; Hollyfield, J.G.; Petrukhin, K. Bestrophin, the product of the Best vitelliform macular dystrophy gene (VMD2), localizes to the basolateral plasma membrane of the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 2000, 97, 12758–12763. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Justus, S.; Li, Y.; Tsang, S.H. BEST1: The Best Target for Gene and Cell Therapies. Mol. Ther. 2015, 23, 1805–1809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Q.; Hartzell, H.C.; Yu, K. Bestrophins and retinopathies. Pflugers Arch. 2010, 460, 559–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boon, C.J.; Klevering, B.J.; Leroy, B.P.; Hoyng, C.B.; Keunen, J.E.; den Hollander, A.I. The spectrum of ocular phenotypes caused by mutations in the BEST1 gene. Prog. Retin Eye Res. 2009, 28, 187–205. [Google Scholar] [CrossRef]
- Best, F., II. Über eine hereditäre Maculaaffektion. Ophthalmologica 1905, 13, 199–212. [Google Scholar] [CrossRef] [Green Version]
- Habibi, I.; Falfoul, Y.; Todorova, M.G.; Wyrsch, S.; Vaclavik, V.; Helfenstein, M.; Turki, A.; Matri, K.E.; Matri, L.E.; Schorderet, D.F. Clinical and Genetic Findings of Autosomal Recessive Bestrophinopathy (ARB). Genes 2019, 10, 953. [Google Scholar] [CrossRef] [Green Version]
- Burgess, R.; Millar, I.D.; Leroy, B.P.; Urquhart, J.E.; Fearon, I.M.; De Baere, E.; Brown, P.D.; Robson, A.G.; Wright, G.A.; Kestelyn, P.; et al. Biallelic mutation of BEST1 causes a distinct retinopathy in humans. Am. J. Hum. Genet. 2008, 82, 19–31. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.S.; Jun, I.; Choi, S.I.; Lee, J.H.; Lee, M.G.; Lee, S.C.; Kim, E.K. A Novel BEST1 Mutation in Autosomal Recessive Bestrophinopathy. Investig. Ophthalmol. Vis. Sci. 2015, 56, 8141–8150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Woo, S.J.; Kim, Y.K.; Lee, S.C.; Lee, C.S. Focal Choroidal Excavation in Multifocal Choroiditis and Punctate Inner Choroidopathy. Ophthalmology 2015, 122, 1534–1535. [Google Scholar] [CrossRef] [PubMed]
- Rim, J.H.; Lee, S.T.; Gee, H.Y.; Lee, B.J.; Choi, J.R.; Park, H.W.; Han, S.H.; Han, J. Accuracy of Next-Generation Sequencing for Molecular Diagnosis in Patients With Infantile Nystagmus Syndrome. JAMA Ophthalmol. 2017, 135, 1376–1385. [Google Scholar] [CrossRef] [PubMed]
- Casalino, G.; Khan, K.N.; Armengol, M.; Wright, G.; Pontikos, N.; Georgiou, M.; Webster, A.R.; Robson, A.G.; Grewal, P.S.; Michaelides, M. Autosomal Recessive Bestrophinopathy: Clinical Features, Natural History, and Genetic Findings in Preparation for Clinical Trials. Ophthalmology 2021, 128, 706–718. [Google Scholar] [CrossRef]
- Gerth, C.; Zawadzki, R.J.; Werner, J.S.; Héon, E. Detailed analysis of retinal function and morphology in a patient with autosomal recessive bestrophinopathy (ARB). Doc. Ophthalmol. 2009, 118, 239–246. [Google Scholar] [CrossRef] [Green Version]
- Wittström, E.; Ekvall, S.; Schatz, P.; Bondeson, M.L.; Ponjavic, V.; Andréasson, S. Morphological and functional changes in multifocal vitelliform retinopathy and biallelic mutations in BEST1. Ophthalmol. Genet. 2011, 32, 83–96. [Google Scholar]
- Zhao, L.; Grob, S.; Corey, R.; Krupa, M.; Luo, J.; Du, H.; Lee, C.; Hughes, G.; Lee, J.; Quach, J.; et al. A novel compound heterozygous mutation in the BEST1 gene causes autosomal recessive Best vitelliform macular dystrophy. Eye 2012, 26, 866–871. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Y.; Xu, Y.; Kittredge, A.; Ward, N.; Chen, S.; Tsang, S.H.; Yang, T. Patient-specific mutations impair BESTROPHIN1’s essential role in mediating Ca2+-dependent Cl- currents in human RPE. Elife 2017, 6, e29914. [Google Scholar] [CrossRef] [Green Version]
- Tian, L.; Sun, T.; Xu, K.; Zhang, X.; Peng, X.; Li, Y. Screening of BEST1 Gene in a Chinese Cohort With Best Vitelliform Macular Dystrophy or Autosomal Recessive Bestrophinopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3366–3375. [Google Scholar] [CrossRef] [Green Version]
- Kinnick, T.R.; Mullins, R.F.; Dev, S.; Leys, M.; Mackey, D.A.; Kay, C.N.; Lam, B.L.; Fishman, G.A.; Traboulsi, E.; Iezzi, R.; et al. Autosomal recessive vitelliform macular dystrophy in a large cohort of vitelliform macular dystrophy patients. Retina 2011, 31, 581–595. [Google Scholar] [CrossRef]
- Bitner, H.; Mizrahi-Meissonnier, L.; Griefner, G.; Erdinest, I.; Sharon, D.; Banin, E. A homozygous frameshift mutation in BEST1 causes the classical form of Best disease in an autosomal recessive mode. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5332–5338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannaccone, A.; Kerr, N.C.; Kinnick, T.R.; Calzada, J.I.; Stone, E.M. Autosomal recessive best vitelliform macular dystrophy: Report of a family and management of early-onset neovascular complications. Arch. Ophthalmol. 2011, 129, 211–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Querques, G.; Bocco, M.C.; Soubrane, G.; Souied, E.H. Intravitreal ranibizumab (Lucentis) for choroidal neovascularization associated with vitelliform macular dystrophy. Acta Ophthalmol. 2008, 86, 694–695. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Patient No. | Sex | Age at Onset (Year) | Age at Diagnosis (Year) | Follow-Up Period (Months) | Visual Acuity * at Initial Visit | Visual Acuity * at Final Visit | Spherical Equivalent | Primary Angle-Closure Glaucoma | Family History | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OD | OS | OD | OS | OD | OS | |||||||
| 1 | F | 33 | 35 | 52 | 0.15 | 0.5 | 0.1 | 0.4 | −2.13 | −1.25 | Yes | Yes (brother) |
| 2 | M | 30 | 33 | 11 | 1.0 | 0.7 | 1.0 | 0.7 | −1.50 | −3.50 | Yes | No |
| 3 | M | 10 | 16 | 14 | 1.0 | 0.6 | 1.0 | 0.5 | 1.50 | 1.88 | No | No |
| 4 | F | 32 | 32 | 44 | 0.7 | 1.0 | 0.5 | 0.5 | −1.00 | −0.88 | Yes | No |
| 5 | F | 20 | 53 | 105 | 0.025 | 0.025 | 0.04 | 0.04 | 4.25 | 4.00 | No | Yes (brother) |
| 6 | M | 25 | 67 | 145 | 0.1 | 0.1 | 0.04 | 0.04 | 2.75 | 2.63 | No | Yes (Sister) |
| 7 | F | 37 | 39 | 21 | 0.5 | 0.8 | 0.4 | 1.0 | −0.63 | −1.75 | No | No |
| 8 | F | 50 | 51 | 10 | 0.2 | 0.2 | 0.15 | 0.15 | −0.50 | 0.13 | No | Suspected (Sister) |
| 9 | M | 15 | 16 | 12 | 0.3 | 0.5 | 0.3 | 0.5 | −1.00 | −0.50 | No | No |
| 10 | F | 6 | 6 | 14 | 0.7 | 0.8 | 0.4 | 0.4 | 2.63 | 3.13 | No | No |
| OCT Parameters | At Initial Visit | At Final Visit |
|---|---|---|
| Macular subretinal deposit (%) | ||
| Unifocal | 30 | 25 |
| Multifocal | 60 | 60 |
| Subretinal fluid (%) | ||
| Sub-foveal | 70 | 60 |
| Diffuse | 10 | 20 |
| Intraretinal fluid (%) | 50 | 40 |
| Focal choroidal excavation (%) | 10 | 10 |
| Central macular thickness (μm) * | 198.8 ± 231.2/173.7 ± 167.4 | 185.9 ± 196.3/153.9 ± 150.7 |
| Subfoveal choroidal thickness (μm) * | 303.4 ± 70.5/280.4 ± 76.7 | 289.3 ± 61.9/286.5 ± 47.4 |
| Outer retinal layer thickening (%) | 70 | 70 |
| Patient No. | BEST1 Mutation | Amino Acid Change | BEST1 Mutation | Amino Acid Change |
|---|---|---|---|---|
| 1 | c.763C>T | p.Arg255Trp | c.113T>G | p.Ile38Ser |
| 2 | c.763C>T | p.Arg255Trp | c.236C>T | p.Ser79Phe |
| 3 | c.584C>T | p.Ala195Val | c.763C>T | p.Arg255Trp |
| 4 | c.452C>T | p.Leu151Pro | c.650C>T | p.Trp217Met |
| 5 | c.119T>C | p.Leu40Pro | c.584C>T | p.Ala195Val |
| 6 | c.119T>C | p.Leu40Pro | c.584C>T | p.Ala195Val |
| 7 | c.584C>T | p.Ala195Val | c.763C>T | p.Arg255Trp |
| 8 | c.584C>T | p.Ala195Val | c.763C>T | p.Arg255Trp |
| 9 | c.140G>A | p.Arg47His | c.113T>G | p.Ile38Ser |
| 10 | c.584C>T | p.Ala195Val | c.632T>C | p.Leu211Pro |
| Patient No. | ERG | EOG (Arden Ratio) | ||
|---|---|---|---|---|
| OD | OS | OD | OS | |
| 1 | WNL | WNL | 1.1 | 1.8 |
| 2 | WNL | WNL | 1.9 | 1.4 |
| 3 | WNL | WNL | 1.8 | 1.5 |
| 4 | WNL | WNL | 1 | 1 |
| 5 | Rod and cone impairment | 1.5 | 2 | |
| 6 | Rod and cone impairment | 0.92 | 1.06 | |
| 7 | Not performed | Not performed | ||
| 8 | Not performed | Not performed | ||
| 9 | WNL | WNL | 1.1 | 1 |
| 10 | Not performed | 1.5 | 1.1 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.R.; Han, J.; Kim, Y.J.; Kang, H.G.; Byeon, S.H.; Kim, S.S.; Lee, C.S. Clinical Features and Genetic Findings of Autosomal Recessive Bestrophinopathy. Genes 2022, 13, 1197. https://doi.org/10.3390/genes13071197
Kim HR, Han J, Kim YJ, Kang HG, Byeon SH, Kim SS, Lee CS. Clinical Features and Genetic Findings of Autosomal Recessive Bestrophinopathy. Genes. 2022; 13(7):1197. https://doi.org/10.3390/genes13071197
Chicago/Turabian StyleKim, Hae Rang, Jinu Han, Yong Joon Kim, Hyun Goo Kang, Suk Ho Byeon, Sung Soo Kim, and Christopher Seungkyu Lee. 2022. "Clinical Features and Genetic Findings of Autosomal Recessive Bestrophinopathy" Genes 13, no. 7: 1197. https://doi.org/10.3390/genes13071197
APA StyleKim, H. R., Han, J., Kim, Y. J., Kang, H. G., Byeon, S. H., Kim, S. S., & Lee, C. S. (2022). Clinical Features and Genetic Findings of Autosomal Recessive Bestrophinopathy. Genes, 13(7), 1197. https://doi.org/10.3390/genes13071197