
A Novel Hypothesis on Choroideremia-Manifesting Female Carriers: Could CHM In-Frame Variants Exert a Dominant Negative Effect? A Case Report

 ,
,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Ophthalmological Assessment
2.3. Genetic Analyses
2.4. Molecular Modelling
3. Results
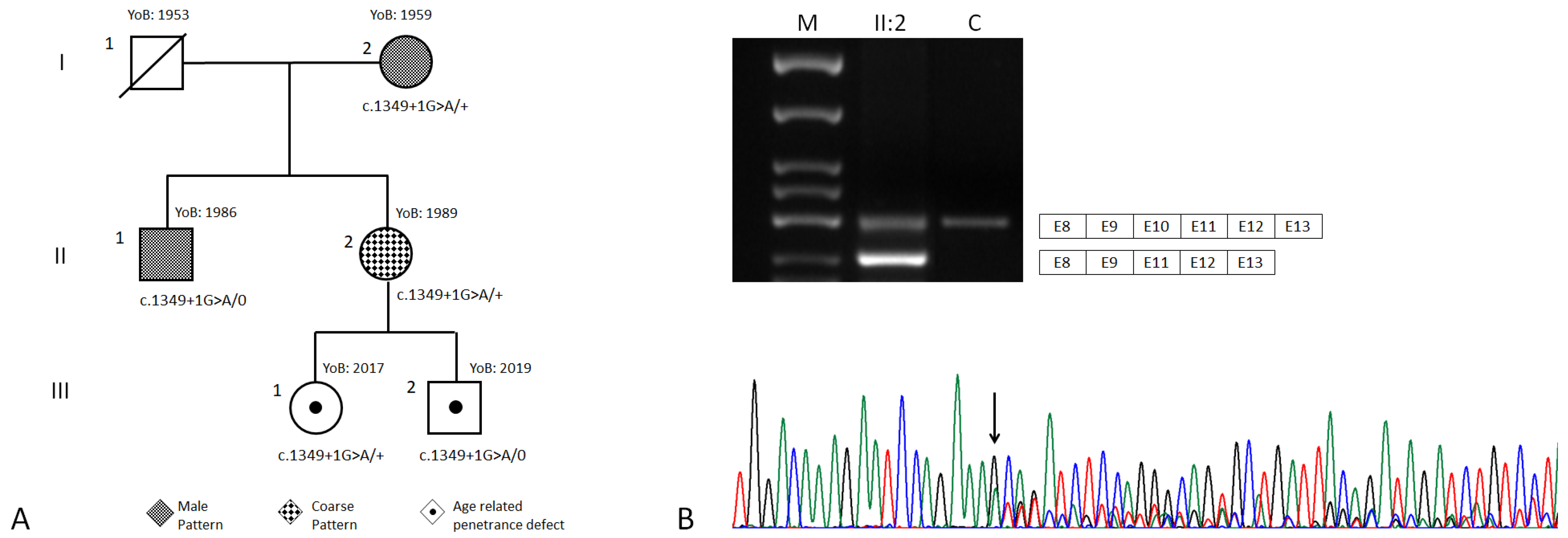
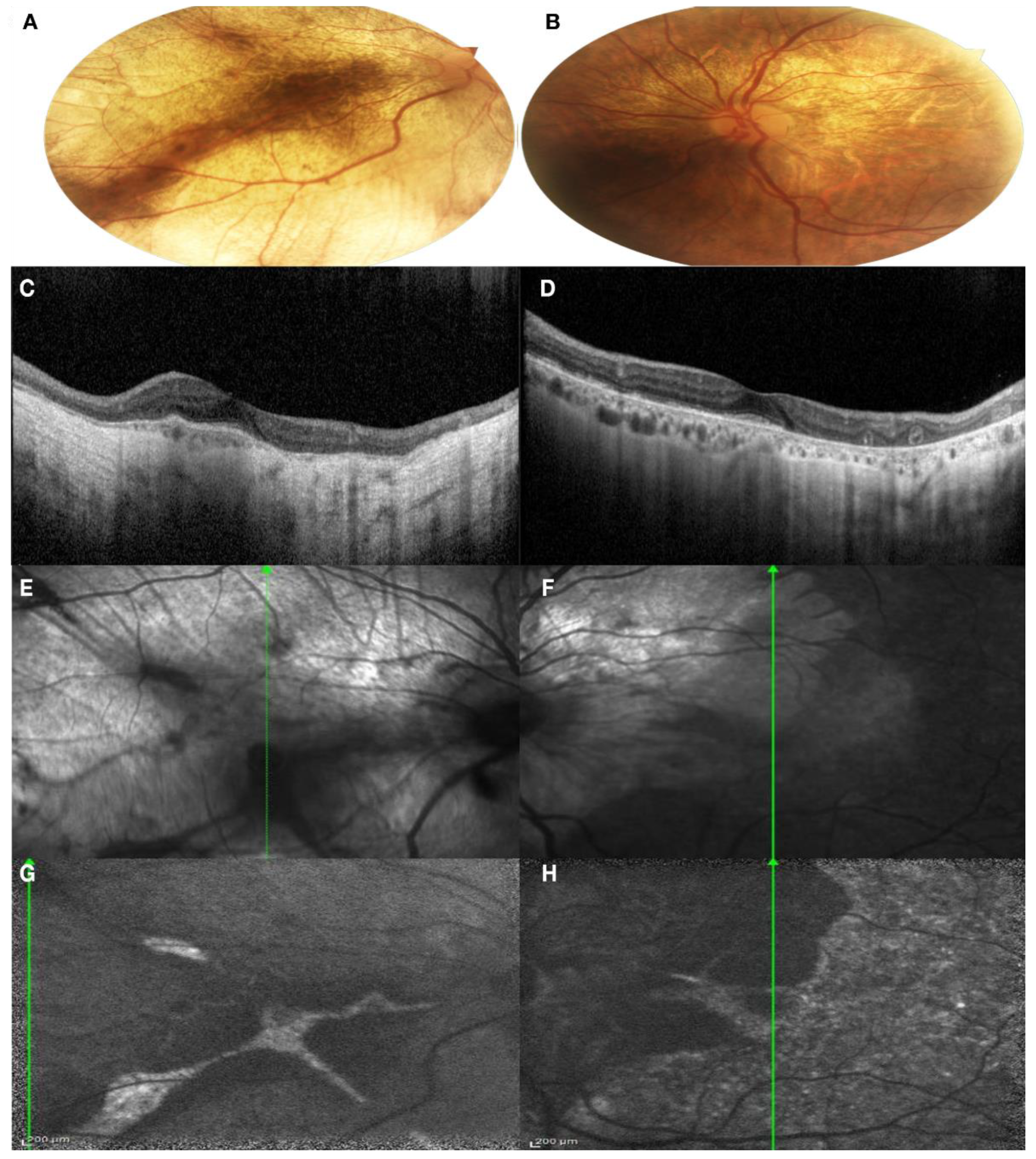
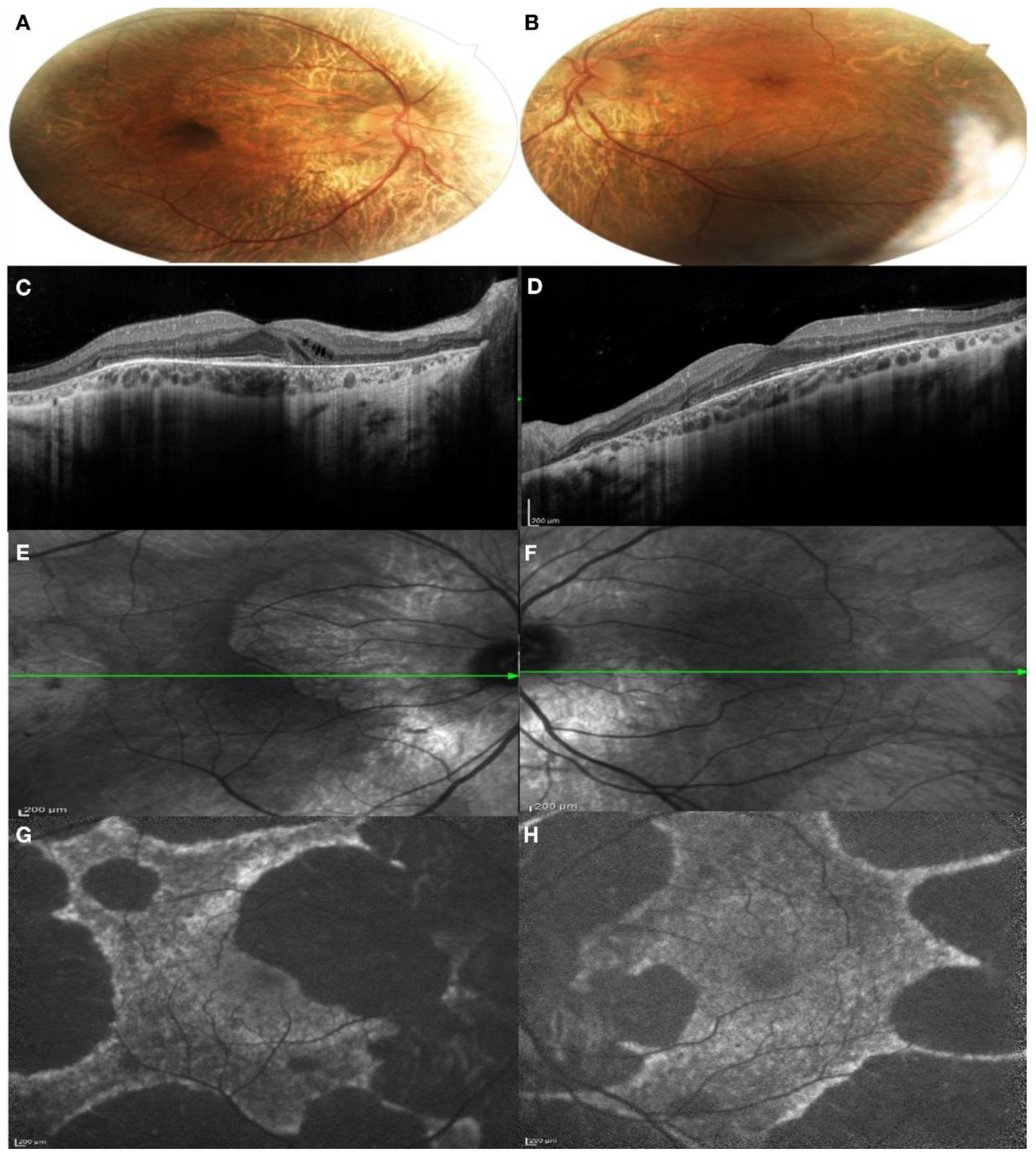
3.1. Clinical Findings
3.2. Genetic Findings
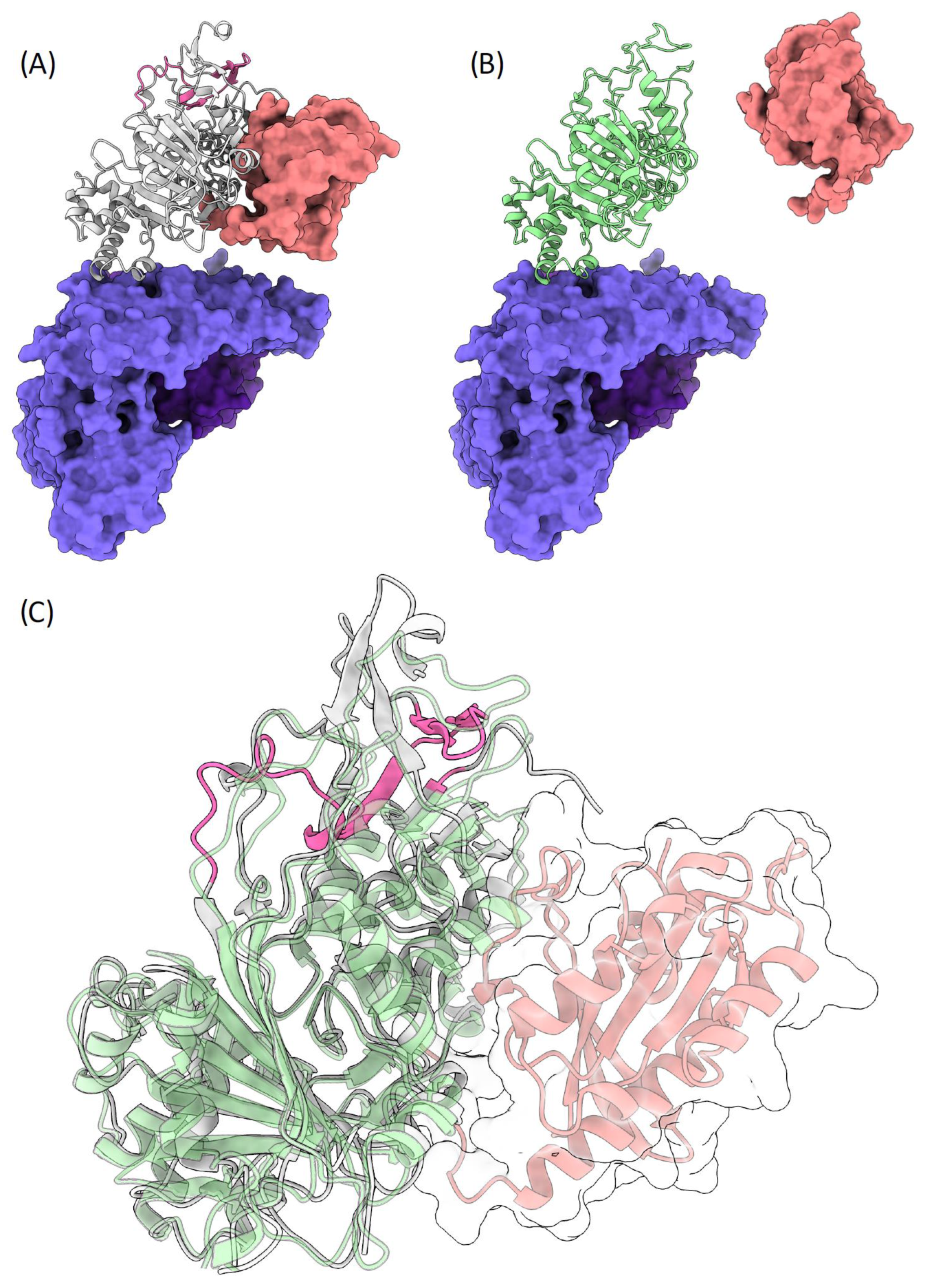
3.3. Molecular Modelling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dimopoulos, I.S.; Chan, S.; MacLaren, R.E.; MacDonald, I.M. Pathogenic mechanisms and the prospect of gene therapy for choroideremia. Expert Opin. Orphan Drugs 2015, 3, 787–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coussa, R.G.; Traboulsi, E.I. Choroideremia: A review of general findings and pathogenesis. Ophthalmic. Genet. 2012, 33, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Zeitz, C.; Nassisi, M.; Laurent-Coriat, C.; Andrieu, C.; Boyard, F.; Condroyer, C.; Démontant, V.; Antonio, A.; Lancelot, M.E.; Frederiksen, H.; et al. CHM mutation spectrum and disease: An update at the time of human therapeutic trials. Hum. Mutat. 2021, 42, 323–341. [Google Scholar] [CrossRef]
- Jauregui, R.; Park, K.S.; Tanaka, A.J.; Cho, A.; Paavo, M.; Zernant, J.; Francis, J.H.; Allikmets, R.; Sparrow, J.R.; Tsang, S.H. Spectrum of Disease Severity and Phenotype in Choroideremia Carriers. Am. J. Ophthalmol. 2019, 207, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.L.; Casey, P.J. Protein prenylation: Molecular mechanisms and functional consequences. Annu. Rev. Biochem. 1996, 65, 241–269. [Google Scholar] [CrossRef]
- Guo, Z.; Wu, Y.W.; Das, D.; Delon, C.; Cramer, J.; Yu, S.; Thuns, S.; Lupilova, N.; Waldmann, H.; Brunsveld, L.; et al. Structures of RabGGTase-substrate/product complexes provide insights into the evolution of protein prenylation. EMBO J. 2008, 27, 2444–2456. [Google Scholar] [CrossRef] [Green Version]
- Baron, R.A.; Seabra, M.C. Rab geranylgeranylation occurs preferentially via the pre-formed REP-RGGT complex and is regulated by geranylgeranyl pyrophosphate. Biochem. J. 2008, 415, 67–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, T.L.; Groppe, M.; Jolly, J.K.; Downes, S.M.; MacLaren, R.E. Correlation of retinal structure and function in choroideremia carriers. Ophthalmology 2015, 122, 1274–1276. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Rak, A.; Pylypenko, O.; Niculae, A.; Pyatkov, K.; Goody, R.S.; Alexandrov, K. Structure of the Rab7:REP-1 complex: Insights into the mechanism of Rab prenylation and choroideremia disease. Cell 2004, 117, 749–760. [Google Scholar] [CrossRef] [Green Version]
- Pylypenko, O.; Rak, A.; Reents, R.; Niculae, A.; Sidorovitch, V.; Cioaca, M.D.; Bessolitsyna, E.; Thomä, N.H.; Waldmann, H.; Schlichting, I.; et al. Structure of Rab escort protein-1 in complex with Rab geranylgeranyltransferase. Mol. Cell 2003, 11, 483–494. [Google Scholar] [CrossRef]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.S.; Eramian, D.; Shen, M.Y.; Pieper, U.; Sali, A. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinform. 2006, 15, 5–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedberg, I.; Kaplan, T.; Margalit, H. Evaluation of PSI-BLAST alignment accuracy in comparison to structural alignments. Protein Sci. 2000, 9, 2278–2284. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janson, G.; Paiardini, A. PyMod 3: A complete suite for structural bioinformatics in PyMOL. Bioinformatics 2021, 37, 1471–1472. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Migeon, B.R. X-linked diseases: Susceptible females. Genet. Med. 2020, 22, 1156–1174. [Google Scholar] [CrossRef] [Green Version]
- Fahim, A.T.; Daiger, S.P. The Role of X-Chromosome Inactivation in Retinal Development and Disease. Adv. Exp. Med. Biol. 2016, 854, 325–331. [Google Scholar]
- Perez-Cano, H.J.; Garnica-Hayashi, R.E.; Zenteno, J.C. CHM gene molecular analysis and X-chromosome inactivation pattern determination in two families with choroideremia. Am. J. Med. Genet. A 2009, 149, 2134–2140. [Google Scholar] [CrossRef]
- Carrel, L.; Willard, H.F. Heterogeneous gene expression from the inactive X chromosome: An X-linked gene that escapes X inactivation in some human cell lines but is inactivated in others. Proc. Natl. Acad. Sci. USA 1999, 96, 7364–7369. [Google Scholar] [CrossRef] [Green Version]
- Carrel, L.; Willard, H.F. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 2005, 434, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Tukiainen, T.; Villani, A.C.; Yen, A.; Rivas, M.A.; Marshall, J.L.; Satija, R.; Aguirre, M.; Gauthier, L.; Fleharty, M.; Kirby, A.; et al. Landscape of X chromosome inactivation across human tissues. Nature 2017, 550, 244–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afanasyeva, T.A.V.; Corral-Serrano, J.C.; Garanto, A.; Roepman, R.; Cheetham, M.E.; Collin, R.W.J. A look into retinal organoids: Methods, analytical techniques, and applications. Cell Mol. Life Sci. 2021, 78, 6505–6532. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Giosaffatte, N.; Valiante, M.; Tricarico, S.; Parise, G.; De Negri, A.M.; Ricciotti, G.; Florean, L.; Paiardini, A.; Bottillo, I.; Grammatico, P. A Novel Hypothesis on Choroideremia-Manifesting Female Carriers: Could CHM In-Frame Variants Exert a Dominant Negative Effect? A Case Report. Genes 2022, 13, 1268. https://doi.org/10.3390/genes13071268
Di Giosaffatte N, Valiante M, Tricarico S, Parise G, De Negri AM, Ricciotti G, Florean L, Paiardini A, Bottillo I, Grammatico P. A Novel Hypothesis on Choroideremia-Manifesting Female Carriers: Could CHM In-Frame Variants Exert a Dominant Negative Effect? A Case Report. Genes. 2022; 13(7):1268. https://doi.org/10.3390/genes13071268
Chicago/Turabian StyleDi Giosaffatte, Niccolò, Michele Valiante, Stefano Tricarico, Giulia Parise, Anna Maria De Negri, Guido Ricciotti, Lara Florean, Alessandro Paiardini, Irene Bottillo, and Paola Grammatico. 2022. "A Novel Hypothesis on Choroideremia-Manifesting Female Carriers: Could CHM In-Frame Variants Exert a Dominant Negative Effect? A Case Report" Genes 13, no. 7: 1268. https://doi.org/10.3390/genes13071268
APA StyleDi Giosaffatte, N., Valiante, M., Tricarico, S., Parise, G., De Negri, A. M., Ricciotti, G., Florean, L., Paiardini, A., Bottillo, I., & Grammatico, P. (2022). A Novel Hypothesis on Choroideremia-Manifesting Female Carriers: Could CHM In-Frame Variants Exert a Dominant Negative Effect? A Case Report. Genes, 13(7), 1268. https://doi.org/10.3390/genes13071268