
DNA Methylation-Specific Analysis of G Protein-Coupled Receptor-Related Genes in Pan-Cancer

Abstract
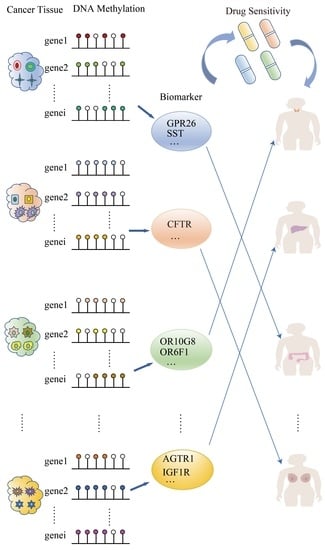
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Dataset and Data Preparation
2.2. Identify Differentially Methylated Sites and Genes
2.3. Functional Annotation of Differentially Methylated Genes
2.4. Feature Selection of Differentially DNA Methylated Sites
2.5. Identification of Pan-Cancer-Specific GPCRs-Related DNA Methylation Genes (GRSDMs)
2.6. Survival Analysis
2.7. Correlation Analysis with Drug Targets
3. Results
3.1. The Landscape of DNA Methylation Genes Related with GPCRs in Pan-Cancer
3.2. Acquisition of Characteristic DNA Methylation Genes in Pan-Cancer
3.3. Identifying Pan-Cancer-Specific GPCRs-Related DNA Methylation Genes (GRSDMs)
3.4. Prognostic Analysis of Specific DNA Methylation Genes
3.5. Validation of Specific DNA Methylation Genes in GEO
3.6. Selection of Potential Drugs Based on Specific DNA Methylation Genes in Pan-Cancer
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wu, L.; Qu, X. Cancer biomarker detection: Recent achievements and challenges. Chem. Soc. Rev. 2015, 44, 2963–2997. [Google Scholar] [CrossRef]
- Hristova, V.A.; Chan, D.W. Cancer biomarker discovery and translation: Proteomics and beyond. Expert Rev. Proteom. 2019, 16, 93–103. [Google Scholar] [CrossRef]
- Huang, H. Matrix Metalloproteinase-9 (MMP-9) as a Cancer Biomarker and MMP-9 Biosensors: Recent Advances. Sensors 2018, 18, 3249. [Google Scholar] [CrossRef] [Green Version]
- Zhan, S.; Yang, P.; Zhou, S.; Xu, Y.; Xu, R.; Liang, G.; Zhang, C.; Chen, X.; Yang, L.; Jin, F.; et al. Serum mitochondrial tsRNA serves as a novel biomarker for hepatocarcinoma diagnosis. Front. Med. 2022, 16, 216–226. [Google Scholar] [CrossRef]
- Pu, Y.; Li, C.; Yuan, H.; Wang, X. Identification of prostate cancer specific methylation biomarkers from a multi-cancer analysis. BMC Bioinform. 2021, 22, 492. [Google Scholar] [CrossRef]
- Dorsam, R.T.; Gutkind, J.S. G-protein-coupled receptors and cancer. Nat. Rev. Cancer 2007, 7, 79–94. [Google Scholar] [CrossRef]
- Whalen, E.; Rajagopal, S.; Lefkowitz, R. Therapeutic potential of β-arrestin- and G protein-biased agonists. Trends Mol. Med. 2011, 17, 126–139. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y. Molecular chaperones and G protein-coupled receptor maturation and pharmacology. Mol. Cell. Endocrinol. 2020, 511, 110862. [Google Scholar] [CrossRef]
- Insel, P.; Sriram, K.; Wiley, S.; Wilderman, A.; Katakia, T.; McCann, T.; Yokouchi, H.; Zhang, L.; Corriden, R.; Liu, D.; et al. GPCRomics: GPCR Expression in Cancer Cells and Tumors Identifies New, Potential Biomarkers and Therapeutic Targets. Front. Pharmacol. 2018, 9, 431. [Google Scholar] [CrossRef] [Green Version]
- Sriram, K.; Moyung, K.; Corriden, R.; Carter, H.; Insel, P.A. GPCRs show widespread differential mRNA expression and frequent mutation and copy number variation in solid tumors. PLoS Biol. 2019, 17, e3000434. [Google Scholar] [CrossRef] [Green Version]
- Byun, S.; Affolter, K.; Snow, A.; Curtin, K.; Cannon, A.; Cannon-Albright, L.; Thota, R.; Neklason, D. Differential methylation of G-protein coupled receptor signaling genes in gastrointestinal neuroendocrine tumors. Sci. Rep. 2021, 11, 12303. [Google Scholar] [CrossRef]
- Gopi, L.K.; Kidder, B.L. Integrative pan cancer analysis reveals epigenomic variation in cancer type and cell specific chromatin domains. Nat. Commun. 2021, 12, 1419. [Google Scholar] [CrossRef]
- Doi, A.; Park, I.H.; Wen, B.; Murakami, P.; Aryee, M.J.; Irizarry, R.; Herb, B.; Ladd-Acosta, C.; Rho, J.; Loewer, S.; et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat. Genet. 2009, 41, 1350–1353. [Google Scholar] [CrossRef] [Green Version]
- Zou, Q.; Wang, X.; Ren, D.; Hu, B.; Tang, G.; Zhang, Y.; Huang, M.; Pai, R.K.; Buchanan, D.D.; Win, A.K.; et al. DNA methylation-based signature of CD8+ tumor-infiltrating lymphocytes enables evaluation of immune response and prognosis in colorectal cancer. J. Immunother. Cancer 2021, 9, e002671. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006, 7, 21–33. [Google Scholar] [CrossRef]
- Issa, J.; Kantarjian, H. Targeting DNA methylation. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 3938–3946. [Google Scholar] [CrossRef] [Green Version]
- Jelinic, P.; Shaw, P. Loss of imprinting and cancer. J. Pathol. 2007, 211, 261–268. [Google Scholar] [CrossRef]
- Jahangiri, R.; Mosaffa, F.; Emami Razavi, A.; Teimoori-Toolabi, L.; Jamialahmadi, K. PAX2 promoter methylation and overexpression promote tamoxifen resistance in breast carcinoma patients. J. Oncol. Pharm. Pract. Off. Publ. Int. Soc. Oncol. Pharm. Pract. 2022, 28, 310–325. [Google Scholar] [CrossRef]
- Abdel-Hafiz, H. Epigenetic Mechanisms of Tamoxifen Resistance in Luminal Breast Cancer. Diseases 2017, 5, 16. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Maeser, D.; Gruener, R.F.; Huang, R.S. oncoPredict: An R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief Bioinform. 2021, 22, bbab260. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.K.; Kim, S. An Insight into GPCR and G-Proteins as Cancer Drivers. Cells 2021, 10, 3288. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Chen, G.; Shi, T. Integrative analysis identifies potential DNA methylation biomarkers for pan-cancer diagnosis and prognosis. Epigenetics 2019, 14, 67–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paziewska, A.; Dabrowska, M.; Goryca, K.; Antoniewicz, A.; Dobruch, J.; Mikula, M.; Jarosz, D.; Zapala, L.; Borowka, A.; Ostrowski, J. DNA methylation status is more reliable than gene expression at detecting cancer in prostate biopsy. Br. J. Cancer 2014, 111, 781–789. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Tsui, S.K.; Wu, H.; Wei, Y. Pan-cancer analysis of differential DNA methylation patterns. BMC Med. Genom. 2020, 13, 154. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, R.; Burwinkel, B.; Breitling, L.P.; Holleczek, B.; Schottker, B.; Brenner, H. F2RL3 methylation in blood DNA is a strong predictor of mortality. Int. J. Epidemiol. 2014, 43, 1215–1225. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, Z.; Liu, R.; Li, Z.; Lin, J.; Wojciechowicz, M.L.; Huang, J.; Lee, K.; Ma’ayan, A.; He, J.C. Connectivity Mapping Identifies BI-2536 as a Potential Drug to Treat Diabetic Kidney Disease. Diabetes 2021, 70, 589–602. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Sosman, J.A.; Quevedo, J.F.; Milhem, M.M.; Joshua, A.M.; Kudchadkar, R.R.; Linette, G.P.; Gajewski, T.F.; Lutzky, J.; Lawson, D.H.; et al. Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma: A randomized clinical trial. JAMA 2014, 311, 2397–2405. [Google Scholar] [CrossRef]
- Amaria, R.N.; Prieto, P.A.; Tetzlaff, M.T.; Reuben, A.; Andrews, M.C.; Ross, M.I.; Glitza, I.C.; Cormier, J.; Hwu, W.J.; Tawbi, H.A.; et al. Neoadjuvant plus adjuvant dabrafenib and trametinib versus standard of care in patients with high-risk, surgically resectable melanoma: A single-centre, open-label, randomised, phase 2 trial. Lancet Oncol. 2018, 19, 181–193. [Google Scholar] [CrossRef]
- Dhillon, S. Dabrafenib plus Trametinib: A Review in Advanced Melanoma with a BRAF (V600) Mutation. Target. Oncol. 2016, 11, 417–428. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Zhao, J.; Dong, H.; Xue, W.; Xing, J.; Liu, T.; Yu, X.; Gu, Y.; Sun, B.; Lu, H.; et al. DNA Methylation-Specific Analysis of G Protein-Coupled Receptor-Related Genes in Pan-Cancer. Genes 2022, 13, 1213. https://doi.org/10.3390/genes13071213
Zhang M, Zhao J, Dong H, Xue W, Xing J, Liu T, Yu X, Gu Y, Sun B, Lu H, et al. DNA Methylation-Specific Analysis of G Protein-Coupled Receptor-Related Genes in Pan-Cancer. Genes. 2022; 13(7):1213. https://doi.org/10.3390/genes13071213
Chicago/Turabian StyleZhang, Mengyan, Jiyun Zhao, Huili Dong, Wenhui Xue, Jie Xing, Ting Liu, Xiuwen Yu, Yue Gu, Baoqing Sun, Haibo Lu, and et al. 2022. "DNA Methylation-Specific Analysis of G Protein-Coupled Receptor-Related Genes in Pan-Cancer" Genes 13, no. 7: 1213. https://doi.org/10.3390/genes13071213