
Peripheral Myelin Protein 22 Gene Mutations in Charcot-Marie-Tooth Disease Type 1E Patients

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Ethics Statements
2.2. Clinical and Electrophysiological Examinations
2.3. DNA Purification and Detection of Mutations
2.4. In Silico Prediction and Conservation Analysis
2.5. Statistical Analysis
3. Results
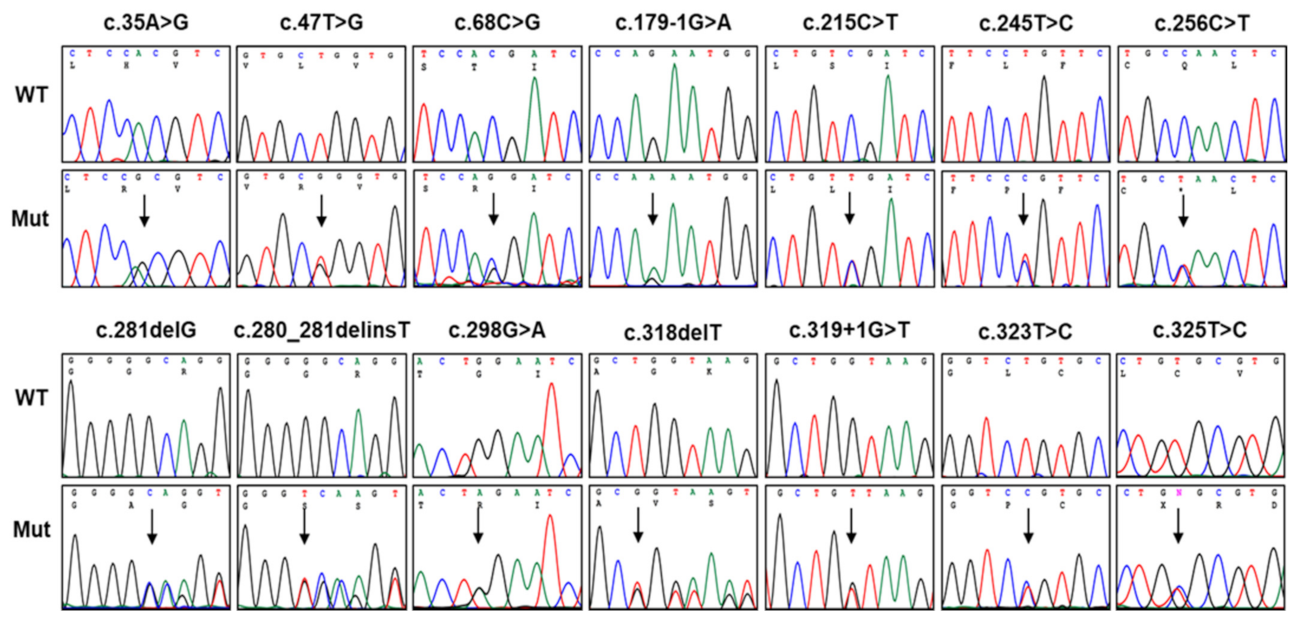
3.1. Identification of Pathogenic Mutations in PMP22
3.2. In Silico Prediction, Conservation, and Conformational Changes
3.3. Mutational Hotspots and an Atypical Case with Somatic Mutation
3.4. Prevalence of CMT1E in CMT Patients
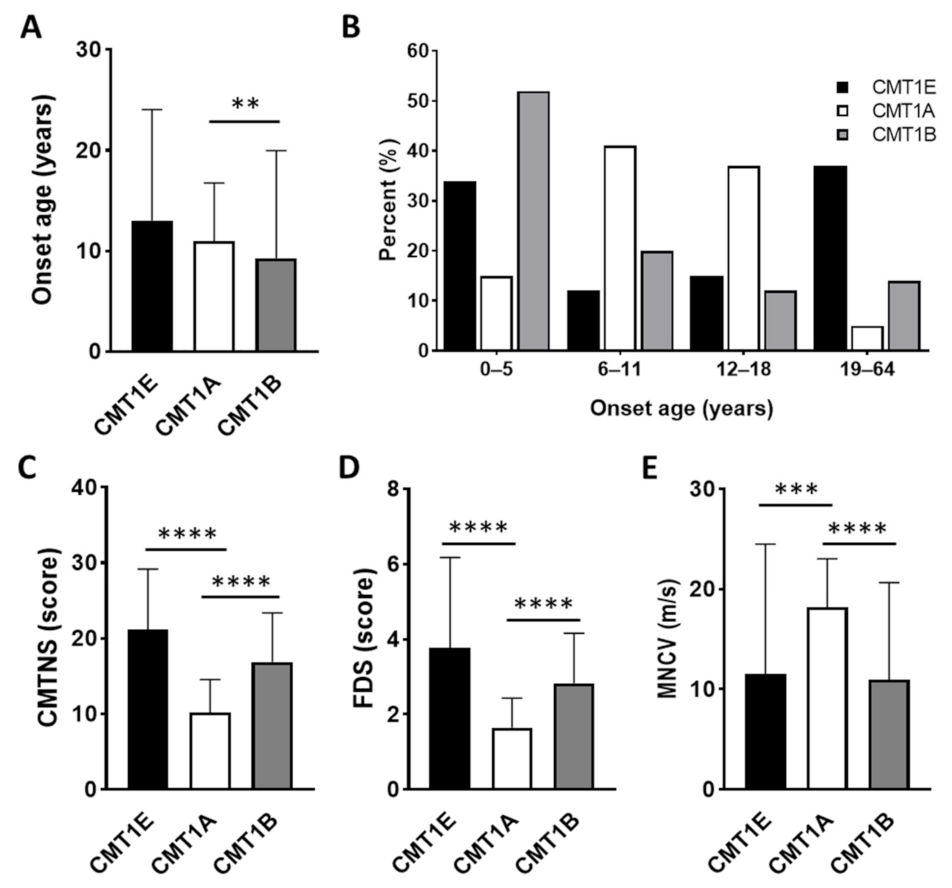
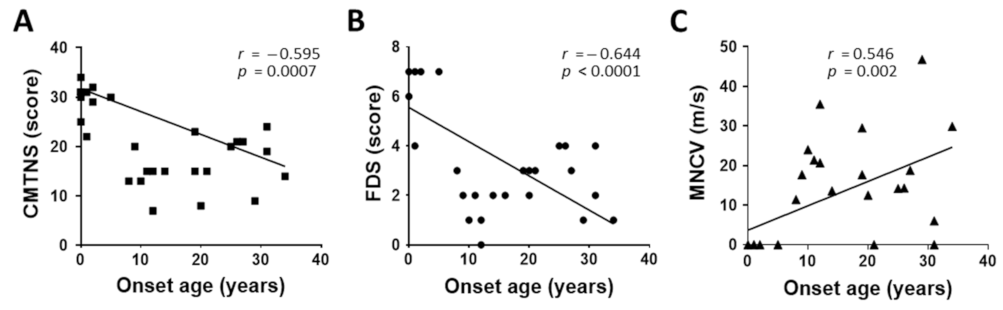
3.5. Characterization of Clinical and Electrophysiological Phenotypes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pareyson, D.; Scaioli, V.; Laurà, M. Clinical and electrophysiological aspects of Charcot-Marie-Tooth disease. Neuromolecular Med. 2006, 8, 3–22. [Google Scholar] [CrossRef]
- Pipis, M.; Rossor, A.M.; Laura, M.; Reilly, M.M. Next-generation sequencing in Charcot-Marie-Tooth disease: Opportunities and challenges. Nat. Rev. Neurol. 2019, 15, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Lupski, J.R.; Montes de Oca-Luna, R.; Slaugenhaupt, S.; Pentao, L.; Guzzetta, V.; Trask, B.J.; Saucedo-Cardenas, O.; Barker, D.F.; Killian, J.M.; Garcia, C.A.; et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell 1991, 66, 219–232. [Google Scholar] [CrossRef]
- Chance, P.F.; Alderson, M.K.; Leppig, K.A.; Lensch, M.W.; Matsunami, N.; Smith, B.; Swanson, P.D.; Odelberg, S.J.; Disteche, C.M.; Bird, T.D. DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell 1993, 72, 143–151. [Google Scholar] [CrossRef]
- Boerkoel, C.F.; Takashima, H.; Garcia, C.A.; Olney, R.K.; Johnson, J.; Berry, K.; Russo, P.; Kennedy, S.; Teebi, A.S.; Scavina, M.; et al. Charcot-Marie-Tooth disease and related neuropathies: Mutation distribution and genotype-phenotype correlation. Ann. Neurol. 2002, 51, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Kovach, M.J.; Lin, J.-P.; Boyadjiev, S.; Campbell, K.; Mazzeo, L.; Herman, K.; Rimer, L.A.; Frank, W.; Llewellyn, B.; Jabs, E.W.; et al. A unique point mutation in the PMP22 gene is associated with Charcot-Marie-Tooth disease and deafness. Am. J. Hum. Genet. 1999, 64, 1580–1593. [Google Scholar] [CrossRef] [Green Version]
- Joo, I.S.; Ki, C.S.; Joo, S.Y.; Huh, K.; Kim, J.W. A novel point mutation in PMP22 gene associated with a familial case of Charcot-Marie-Tooth disease type 1A with sensorineural deafness. Neuromuscul. Disord. 2004, 14, 325–328. [Google Scholar] [CrossRef]
- Kleopa, K.A.; Georgiou, D.-M.; Nicolaou, P.; Koutsou, P.; Papathanasiou, E.; Kyriakides, T.; Christodoulou, K. A novel PMP22 mutation ser22phe in a family with hereditary neuropathy with liability to pressure palsies and CMT1A phenotypes. Neurogenetics 2004, 5, 171–175. [Google Scholar] [CrossRef]
- Valentijn, L.J.; Ouvrier, R.A.; van den Bosch, N.H.A.; Bolhuis, P.A.; Baas, F.; Nicholson, G.A. Dejerine-Sottas neuropathy is associated with a de novo PMP22 mutation. Hum. Mutat. 1995, 5, 76–80. [Google Scholar] [CrossRef]
- Roa, B.B.; Dyck, P.J.; Marks, H.G.; Chance, P.F.; Lupski, J.R. Dejerine-Sottas syndrome associated with point mutation in the peripheral myelin protein 22 (PMP22) gene. Nat. Genet. 1993, 5, 269–273. [Google Scholar] [CrossRef]
- Ionasescu, V.V.; Searby, C.; Greenberg, S.A. Dejerine-Sottas disease with sensorineural hearing loss, nystagmus, and peripheral facial nerve weakness: De novo dominant point mutation of the PMP22 gene. J. Med. Genet. 1996, 33, 1048–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ionasescu, V.V.; Searby, C.C.; Ionasescu, R.; Chatkupt, S.; Patel, N.; Koenigsberger, R. Dejerine-Sottas neuropathy in mother and son with same point mutation of PMP22 gene. Muscle Nerve 1997, 20, 97–99. [Google Scholar] [CrossRef]
- Parman, Y.; Plante-Bordeneuve, V.; Guiochon-Mantel, A.; Eraksoy, M.; Said, G. Recessive inheritance of a new point mutation of the PMP22 gene in Dejerine-Sottas disease. Ann. Neurol. 1999, 45, 518–522. [Google Scholar] [CrossRef]
- Korn-Lubetzki, I.; Argov, Z.; Raas-Rothschild, A.; Wirguin, I.; Steiner, I. Family with inflammatory demyelinating polyneuropathy and the HNPP 17p12 deletion. Am. J. Med. Genet. 2002, 113, 275–278. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Parker, B.; Martyn, C.; Natarajan, C.; Guo, J. The PMP22 gene and its related diseases. Mol. Neurobiol. 2013, 47, 673––698. [Google Scholar] [CrossRef] [Green Version]
- Roa, B.B.; Garcia, C.A.; Pentao, L.; Killian, J.M.; Trask, B.J.; Suter, U.; Snipes, G.J.; Ortiz-Lopez, R.; Shooter, E.M.; Patel, P.I.; et al. Evidence for a recessive PMP22 point mutation in Charcot-Marie-Tooth disease type 1A. Nat. Genet. 1993, 5, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Watila, M.M.; Balarabe, S.A. Molecular and clinical features of inherited neuropathies due to PMP22 duplication. J. Neurol. Sci. 2015, 355, 18–24. [Google Scholar] [CrossRef]
- Sereda, M.; Griffiths, I.; Puhlhofer, A.; Stewart, H.; Rossner, M.J.; Zimmermann, F.; Magyar, J.P.; Schneider, A.; Hund, E.; Meinck, H.M.; et al. A transgenic rat model of Charcot-Marie-Tooth disease. Neuron 1996, 16, 1049–1060. [Google Scholar] [CrossRef] [Green Version]
- Tobler, A.R.; Liu, N.; Mueller, L.; Shooter, E.M. Differential aggregation of the Trembler and Trembler J mutants of peripheral myelin protein 22. Proc. Nat. Acad. Sci. USA 2002, 99, 483–488. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.O.; Lee, M.S.; Shin, S.H.; Hwang, J.H.; Choi, K.G.; Kim, W.K.; Sunwoo, I.N.; Kim, N.K.; Chung, K.W. Mutational analysis of PMP22, MPZ, GJB1, EGR2 and NEFL in Korean Charcot-Marie-Tooth neuropathy patients. Hum. Mutat. 2004, 24, 185–186. [Google Scholar] [CrossRef]
- Choi, B.O.; Koo, S.K.; Park, M.H.; Rhee, H.; Yang, S.J.; Choi, K.G.; Jung, S.C.; Kim, H.S.; Hyun, Y.S.; Nakhro, K.; et al. Exome sequencing is an efficient tool for genetic screening of Charcot-Marie-Tooth disease. Hum. Mutat. 2012, 33, 1610–1615. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.H.; Hong, Y.B.; Hyun, Y.S.; Nam, D.E.; Kwak, G.; Hwang, S.H.; Choi, B.O.; Chung, K.W. Identification of genetic causes of inherited peripheral neuropathies by targeted gene panel sequencing. Mol. Cells 2016, 39, 382–388. [Google Scholar] [PubMed] [Green Version]
- Kim, J.Y.; Koo, H.; Park, K.D.; Choi, S.S.; Yu, J.S.; Hong, Y.B.; Chung, K.W.; Choi, B.O. Genotype–phenotype correlation of Charcot-Marie-Tooth type 1E patients with PMP22 mutations. Genes Genom. 2016, 38, 659–667. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, S.H.; Park, J.Y.; Koo, H.; Park, K.D.; Hong, Y.B.; Chung, K.W.; Chot, B.O. A longitudinal clinicopathological study of two unrelated patients with Charcot-Marie-Tooth disease type 1E. Neurol. India 2017, 65, 893–895. [Google Scholar] [PubMed]
- Park, J.; Kim, H.S.; Kwon, H.M.; Kim, J.; Nam, S.H.; Jung, N.Y.; Lee, A.J.; Jung, Y.H.; Kim, S.B.; Chung, K.W.; et al. Identification and clinical characterization of Charcot-Marie-Tooth disease type 1C patients with LITAF p.G112S mutation. Genes Genom. 2022, in press. [Google Scholar] [CrossRef]
- Murphy, S.M.; Herrmann, D.N.; McDermott, M.P.; Scherer, S.S.; Shy, M.E.; Reilly, M.M.; Pareyson, D. Reliability of the CMT neuropathy score (second version) in Charcot-Marie-Tooth disease. J. Peripher. Nerv. Syst. 2011, 16, 191–198. [Google Scholar] [CrossRef] [Green Version]
- Sadjadi, R.; Reilly, M.M.; Shy, M.E.; Pareyson, D.; Laura, M.; Murphy, S.; Feely, S.M.; Grider, T.; Bacon, C.; Piscosquito, G.; et al. Psychometrics evaluation of Charcot-Marie-Tooth Neuropathy Score (CMTNSv2) second version, using Rasch analysis. J. Peripher. Nerv. Syst. 2014, 19, 192–196. [Google Scholar] [CrossRef] [Green Version]
- Fridman, V.; Sillau, S.; Acsadi, G.; Bacon, C.; Dooley, K.; Burns, J.; Day, J.; Feely, S.; Finkel, R.S.; Grider, T.; et al. Inherited neuropathies consortium-rare diseases clinical research, a longitudinal study of CMT1A using Rasch analysis based CMT neuropathy and examination scores. Neurology 2020, 94, e884–e896. [Google Scholar] [CrossRef]
- Calia, L.; Marques, W., Jr.; Gouvea, S.P.; Lourenço, C.M.; de Oliveira, A.S. Proptosis in a family with the p16 Leuc-to-Prol mutation in the PMP22 gene (CMT 1E). Arq. Neuropsiquiatr. 2013, 71, 332––333. [Google Scholar] [CrossRef] [Green Version]
- Luigetti, M.; Zollino, M.; Conti, G.; Romano, A.; Sabatelli, M. Inherited neuropathies and deafness caused by a PMP22 point mutation: A case report and a review of the literature. Neurol. Sci. 2013, 34, 1705–1707. [Google Scholar] [CrossRef]
- Meuleman, J.; Pou-Serradell, A.; Löfgren, A.; Ceuterick, C.; Martin, J.J.; Timmerman, V.; Van Broeckhoven, C.; De Jonghe, P. A novel 3’-splice site mutation in peripheral myelin protein 22 causing hereditary neuropathy with liability to pressure palsies. Neuromuscul. Disord. 2001, 11, 400–403. [Google Scholar] [CrossRef]
- Simonati, A.; Fabrizi, G.M.; Pasquinelli, A.; Taioli, F.; Cavallaro, T.; Morbin, M.; Marcon, G.; Papini, M.; Rizzuto, N. Congenital hypomyelination neuropathy with Ser72Leu substitution in PMP22. Neuromuscul. Disord. 1999, 9, 257–261. [Google Scholar] [CrossRef]
- Numakura, C.; Lin, C.; Ikegami, T.; Guldberg, P.; Hayasaka, K. Molecular analysis in Japanese patients with Charcot-Marie-Tooth disease: DGGE analysis for PMP22, MPZ, and Cx32/GJB1 mutations. Hum. Mutat. 2002, 20, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.C.; Tsai, P.C.; Lin, T.S.; Hsiao, C.T.; Chao, N.C.; Lin, K.P.; Lee, Y.C. Clinical and molecular characterization of PMP22 point mutations in Taiwanese patients with inherited neuropathy. Sci. Rep. 2017, 7, 15363. [Google Scholar] [CrossRef] [Green Version]
- Takashima, H.; Boerkoel, C.F.; Lupski, J.R. Screening for mutations in a genetically heterogeneous disorder: DHPLC versus DNA sequence for mutation detection in multiple genes causing Charcot-Marie-Tooth neuropathy. Genet. Med. 2001, 3, 335–342. [Google Scholar] [CrossRef] [Green Version]
- Bort, S.; Nelis, E.; Timmerman, V.; Sevilla, T.; Cruz-Martínez, A.; Martínez, F.; Millán, J.M.; Arpa, J.; Vílchez, J.J.; Prieto, F.; et al. Mutational analysis of the MPZ, PMP22 and Cx32 genes in patients of Spanish ancestry with Charcot-Marie-Tooth disease and hereditary neuropathy with liability to pressure palsies. Hum. Genet. 1997, 99, 746–754. [Google Scholar] [CrossRef]
- Zubair, S.; Holland, N.R.; Beson, B.; Parke, J.T.; Prodan, C.I. A novel point mutation in the PMP22 gene in a family with Roussy-Levy syndrome. J Neurol 2008, 255, 1417–1418. [Google Scholar] [CrossRef]
- Hsu, Y.H.; Lin, K.P.; Guo, Y.C.; Tsai, Y.S.; Liao, Y.C.; Lee, Y.C. Mutation spectrum of Charcot-Marie-Tooth disease among the Han Chinese in Taiwan. Ann. Clin. Transl. Neurol. 2019, 6, 1090–1101. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Lin, Z.; Liu, L.; Li, X.; Huang, S.; Zhao, H.; Wang, B.; Zeng, S.; Cao, W.; Li, L.; et al. Genotype and phenotype distribution of 435 patients with Charcot-Marie-Tooth disease from central south China. Eur. J. Neurol. 2021, 28, 3774–3783. [Google Scholar] [CrossRef]
- Gentile, L.; Russo, M.; Fabrizi, G.M.; Taioli, F.; Ferrarini, M.; Testi, S.; Alfonzo, A.; Aguennouz, M.; Toscano, A.; Vita, G.; et al. Charcot-Marie-Tooth disease: Experience from a large Italian tertiary neuromuscular center. Neurol. Sci. 2020, 41, 1239–1243. [Google Scholar] [CrossRef]
- Uchôa Cavalcanti, E.B.; Santos, S.C.L.; Martins, C.E.S.; de Carvalho, D.R.; Rizzo, I.M.P.O.; Freitas, M.C.D.N.B.; da Silva Freitas, D.; de Souza, F.S.; Junior, A.M.; do Nascimento, O.J.M. Charcot-Marie-Tooth disease: Genetic profile of patients from a large Brazilian neuromuscular reference center. J. Peripher. Nerv. Syst. 2021, 26, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Milley, G.M.; Varga, E.T.; Grosz, Z.; Nemes, C.; Arányi, Z.; Boczán, J.; Diószeghy, P.; Molnár, M.J.; Gál, A. Genotypic and phenotypic spectrum of the most common causative genes of Charcot-Marie-Tooth disease in Hungarian patients. Neuromuscul. Disord. 2018, 28, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Sivera, R.; Sevilla, T.; Vílchez, J.J.; Martínez-Rubio, D.; Chumillas, M.J.; Vázquez, J.F.; Muelas, N.; Bataller, L.; Millán, J.M.; Palau, F.; et al. Charcot-Marie-Tooth disease: Genetic and clinical spectrum in a Spanish clinical series. Neurology 2013, 81, 1617–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimura, A.; Yuan, J.H.; Hashiguchi, A.; Ando, M.; Higuchi, Y.; Nakamura, T.; Okamoto, Y.; Nakagawa, M.; Takashima, H. Genetic profile and onset features of 1005 patients with Charcot-Marie-Tooth disease in Japan. J. Neurol. Neurosurg. Psychiatry 2019, 90, 195–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higuchi, Y.; Takashima, H. Clinical genetics of Charcot-Marie-Tooth disease. J. Hum. Genet. 2022, in press. [CrossRef]
- Lee, A.J.; Nam, D.E.; Choi, Y.J.; Noh, S.W.; Nam, S.H.; Lee, H.J.; Kim, S.J.; Song, G.J.; Choi, B.O.; Chung, K.W. Paternal gender specificity and mild phenotypes in Charcot-Marie-Tooth type 1A patients with de novo 17p12 rearrangements. Mol. Genet. Genom. Med. 2020, 8, e1380. [Google Scholar] [CrossRef]
- Kim, H.J.; Nam, S.H.; Kwon, H.M.; Lim, S.O.; Park, J.H.; Kim, H.S.; Kim, S.B.; Lee, K.S.; Lee, J.E.; Choi, B.O.; et al. Genetic and clinical spectrums in Korean Charcot-Marie-Tooth disease patients with myelin protein zero mutations. Mol. Genet. Genom. Med. 2021, 9, e1678. [Google Scholar] [CrossRef]
- Lim, S.O.; Jung, N.Y.; Lee, A.J.; Choi, H.J.; Kwon, H.M.; Son, W.; Nam, S.H.; Choi, B.O.; Chung, K.W. Genetic and clinical studies of peripheral neuropathies with three small heat shock protein gene variants in Korea. Genes 2022, 13, 462. [Google Scholar] [CrossRef]
- Nam, D.E.; Park, J.H.; Park, C.E.; Jung, N.Y.; Nam, S.H.; Kwon, H.M.; Kim, H.S.; Kim, S.B.; Son, W.S.; Choi, B.O.; et al. Variants of aminoacyl-tRNA synthetase genes in Charcot-Marie-Tooth disease: A Korean cohort study. J. Peripher. Nerv. Syst. 2022, 27, 38–49. [Google Scholar] [CrossRef]
- Chung, K.W.; Kim, S.B.; Park, K.D.; Choi, K.G.; Lee, J.H.; Eun, H.W.; Suh, J.S.; Hwang, J.H.; Kim, W.K.; Seo, B.C.; et al. Early onset severe and late-onset mild Charcot-Marie-Tooth disease with mitofusin 2 (MFN2) mutations. Brain 2006, 129, 2103–2118. [Google Scholar] [CrossRef]
- Bernard, R.; Boyer, A.; Nègre, P.; Malzac, P.; Latour, P.; Vandenberghe, A.; Philip, N.; Lévy, N. Prenatal detection of the 17p11.2 duplication in Charcot-Marie-Tooth disease type 1A: Necessity of a multidisciplinary approach for heterogeneous disorders. Eur. J. Hum. Genet. 2002, 10, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Passage, E.; Norreel, J.C.; Noack-Fraissignes, P.; Sanguedolce, V.; Pizant, J.; Thirion, X.; Robaglia-Schlupp, A.; Pellissier, J.F.; Fontés, M. Ascorbic acid treatment corrects the phenotype of a mouse model of Charcot-Marie-Tooth disease. Nat. Med. 2004, 10, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Sereda, M.W.; Meyer zu Hörste, G.; Suter, U.; Uzma, N.; Nave, K.A. Therapeutic administration of progesterone antagonist in a model of Charcot-Marie-Tooth disease (CMT-1A). Nat. Med. 2003, 9, 1533–1537. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Chang, E.H.; Koo, O.J.; Jwa, D.H.; Mo, W.M.; Kwak, G.; Moon, H.W.; Park, H.T.; Hong, Y.B.; Choi, B.O. Pmp22 mutant allele-specific siRNA alleviates demyelinating neuropathic phenotype in vivo. Neurobiol. Dis. 2017, 100, 99–107. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutations 1 | No. of Families | Family IDs | Mutant Allele Frequencies 2 | ACMG-AMP | Notes and References | |||
|---|---|---|---|---|---|---|---|---|
| Nucleotide | Amino Acid | 1000G | gnomAD | KRGDB | ||||
| c.35A>G | p.H12R | 1 | FC618 | NR | NR | NR | P | Novel, de novo |
| c.47T>G | p.L16R | 1 | FC541 | NR | NR | NR | LP | [22] |
| c.68C>G | p.T23R | 3 | FC50, FC303, FC680 | NR | NR | NR | P | [7,30] |
| c.179-1G>A | Splicing site | 2 | FC970 | NR | NR | NR | LP | Novel |
| c.215C>T | p.S72L | 5 | FC285, FC376, FC732, FC829, FC895 | NR | NR | NR | P | [10,11,21,23,24,32], de novo: FC285, FC376, FC732, FC829 |
| c.245T>C | p.L82P | 1 | FC416 | NR | NR | NR | LP | [23], de novo |
| c.256C>T | p.Q86X | 2 | FC1102, FC1325 | NR | NR | NR | P | [33,34] |
| c.281delG | p.G94Afs*16 | 1 | FC1061 | NR | NR | NR | P | [12,35], de novo |
| c.280_281delinsT | p.G94Sfs*16 | 1 | FC1088 | NR | NR | NR | LP | Novel |
| c.298G>A | p.G100R | 1 | FC1140 | NR | NR | NR | P | [36], de novo |
| c.318delT | p.G107Vfs*3 | 1 | FC35 | NR | NR | NR | P | [20] |
| c.319+1G>T | Splicing site | 1 | FC608 | NR | NR | NR | P | Novel |
| c.323T>C | p.L108P | 1 | FC1060 | NR | NR | NR | P | [37] |
| c.325T>C | p.C109R | 1 | FC284 | NR | NR | NR | P | [21,24], de novo |
| Populations | Examined Numbers | Number of CMT1E Patients | Prevalence (%) | References | ||
|---|---|---|---|---|---|---|
| Total | CMT1A Exclusion | Total | CMT1A Exclusion | |||
| Korean | 1243 | 850 | 21 | 1.69 | 2.47 | This study |
| South Chinese | 421 | 336 | 10 | 2.38 | 2.98 | [39] |
| Chinese (Taiwan) | 427 | 219 | 4 | 0.94 | 1.83 | [38] |
| Japanese | - | 2598 | 21 | - | 0.81 | [45] |
| Japanese | - | 1005 | 13 | - | 1.29 | [44] |
| Italian | 295 | 238 | 4 | 1.36 | 1.68 | [40] |
| Brazilian | 286 | 81 | 6 | 2.10 | 7.41 | [41] |
| Hungarian | 531 | 320 | 2 | 0.37 | 0.63 | [42] |
| Spanish | 438 | 254 | 2 | 0.46 | 0.79 | [43] |
| Patient ID | Sex | Age (yrs) | CMTNS | CMTES | FDS | MRC 1 | DTR 2 Knee/Ankle | HL | Median Nerve Conduction Study | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Exam. | Onset | UEx | LEx | CMAP (mV) | MNCV (m/s) | SNAP (μV) | SNCV (m/s) | |||||||
| FC618 (II-1) | M | 1 | 1 | 31 | 23 | 7 | +++ | +++ | A/A | No | A | A | A | A |
| FC541 | M | 13 | 12 | ND | ND | 1 | + | ++ | A/A | Yes | ND | ND | ND | ND |
| FC50 (II-2) | F | 84 | 31 | 24 | 20 | 4 | ++ | ++ | A/A | Yes | A | A | A | A |
| FC50 (III-7) | F | 64 | 25 | 20 | 16 | 4 | ++ | +++ | A/A | Yes | 0.7 | 14.2 | A | A |
| FC50 (III-8) | M | 49 | 19 | 23 | 18 | 3 | ++ | ++ | A/A | Yes | 1.2 | 17.7 | A | A |
| FC50 (III-11) | F | 45 | 26 | 21 | 17 | 4 | ++ | +++ | A/A | Yes | 4.1 | 14.3 | A | A |
| FC50 (IV-8) | M | 20 | 12 | 15 | 11 | 1 | + | ++ | A/A | Yes | 6.1 | 20.7 | A | A |
| FC50 (IV-11) | M | 15 | 11 | 15 | 11 | 2 | + | ++ | A/A | Yes | 8.9 | 21.4 | A | A |
| FC303 | M | 20 | 10 | 13 | 9 | 1 | + | + | D/A | No | 11.6 | 24 | A | A |
| FC680 (I-1) | M | 70 | 27 | 21 | 17 | 3 | ++ | ++ | A/A | Yes | 8.1 | 18.8 | 2.4 | 18.2 |
| FC680 (II-1) | M | 34 | 9 | 20 | 16 | 2 | + | + | A/A | Yes | 3.3 | 17.7 | A | A |
| FC970 (II-3) | F | 40 | 29 | 9 | 9 | 1 | + | + | D/D | No | 19.0 | 46.8 | 28.6 | 35.3 |
| FC970 (II-4) | F | 39 | 34 | 14 | 11 | 1 | + | + | D/D | Yes | 12.3 | 29.9 | 9.4 | 26.2 |
| FC970 (III-1) | F | 13 | 12 | 7 | 6 | 0 | - | + | N/D | No | 17.4 | 35.5 | 20.9 | 29.2 |
| FC285 (II-1) | M | 16 | <1 | 31 | 23 | 7 | +++ | +++ | A/A | No | A | A | A | A |
| FC376 (II-1) | F | 11 | 2 | 32 | 24 | 7 | +++ | +++ | A/A | No | A | A | A | A |
| FC732 (II-2) | F | 7 | 2 | 29 | 21 | 7 | ++ | +++ | A/A | No | A | A | A | A |
| FC829 (III-3) | F | 12 | <1 | 25 | 17 | 7 | ++ | +++ | A/A | No | A | A | 1.8 | 20.8 |
| FC895 | M | 7 | <1 | 31 | 23 | 6 | +++ | +++ | A/A | No | A | A | A | A |
| FC416 (II-2) | F | 28 | <1 | 30 | 22 | 7 | ++ | +++ | A/A | No | A | A | A | A |
| FC1102 (I-2) | F | 47 | 16 | ND | ND | 2 | + | ++ | D/A | No | ND | ND | ND | ND |
| FC1102 (II-1) | M | 21 | 14 | 15 | 11 | 2 | + | ++ | D/A | No | 6.3 | 13.6 | A | A |
| FC1325 (II-3) | M | 54 | 8 | 13 | 15 | 3 | + | ++ | A/A | Yes | 1.8 | 11.4 | A | A |
| FC1325 (III-1) | M | 31 | 20 | 8 | 11 | 2 | + | + | D/A | No | 6.8 | 12.5 | A | A |
| FC1061 (III-1) | F | 26 | <1 | 31 | 23 | 7 | +++ | +++ | A/A | No | A | A | A | A |
| FC1088 | F | 48 | 5 | 30 | 22 | 7 | +++ | +++ | A/A | No | A | A | A | A |
| FC1140 (II-2) | F | 6 | 1 | 22 | 16 | 4 | ++ | +++ | A/A | No | A | A | A | A |
| FC35 (II-5) | M | 34 | 20 | ND | ND | 3 | + | ++ | A/A | Yes | ND | ND | ND | ND |
| FC35 (II-6) | M | 32 | 19 | 15 | 12 | 3 | + | ++ | A/A | Yes | 10.0 | 29.5 | A | A |
| FC608 | M | 39 | 31 | 19 | 12 | 2 | + | ++ | A/A | No | 0.5 | 6.0 | A | A |
| FC1060 | M | 33 | 21 | 15 | 9 | 3 | + | ++ | D/A | No | A | A | A | A |
| FC284 (II-1) | F | 15 | <1 | 34 | 26 | 7 | +++ | +++ | A/A | No | A | A | A | A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, N.Y.; Kwon, H.M.; Nam, D.E.; Tamanna, N.; Lee, A.J.; Kim, S.B.; Choi, B.-O.; Chung, K.W. Peripheral Myelin Protein 22 Gene Mutations in Charcot-Marie-Tooth Disease Type 1E Patients. Genes 2022, 13, 1219. https://doi.org/10.3390/genes13071219
Jung NY, Kwon HM, Nam DE, Tamanna N, Lee AJ, Kim SB, Choi B-O, Chung KW. Peripheral Myelin Protein 22 Gene Mutations in Charcot-Marie-Tooth Disease Type 1E Patients. Genes. 2022; 13(7):1219. https://doi.org/10.3390/genes13071219
Chicago/Turabian StyleJung, Na Young, Hye Mi Kwon, Da Eun Nam, Nasrin Tamanna, Ah Jin Lee, Sang Beom Kim, Byung-Ok Choi, and Ki Wha Chung. 2022. "Peripheral Myelin Protein 22 Gene Mutations in Charcot-Marie-Tooth Disease Type 1E Patients" Genes 13, no. 7: 1219. https://doi.org/10.3390/genes13071219