
Dynamics of the Gut Microbiome and Transcriptome in Korea Native Ricefish (Oryzias latipes) during Chronic Antibiotic Exposure

 , and
, and
Abstract
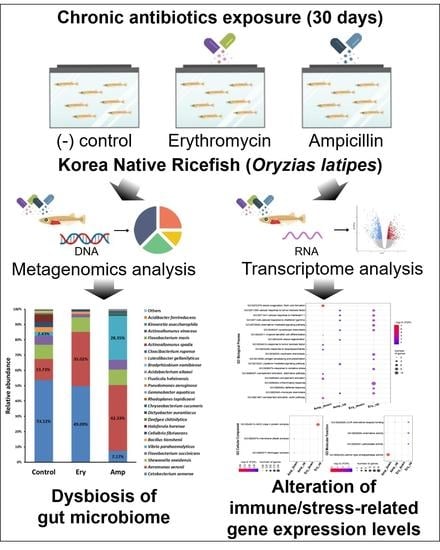
:
1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.2. Chronic Antibiotics Exposure Assay
2.3. Gut Microbe Isolation and 16S rDNA Sequencing
2.4. Minimum Inhibitory Concentration (MIC) Test against O. latipes Gut Isolates
2.5. O. latipes Gut Microbiome Analysis
2.6. Differentially Expressed Genes (DEGs) Analysis
2.7. Statistical Analysis
3. Results
3.1. Determination of Exposure Concentration of Ampicillin and Erythromycin in O. latipes Gut Isolates
3.2. Effects of Chronic Antibiotic Exposure on the Richness and Diversity of the Gut Microbiome
3.3. Effect of Chronic Exposure to Antibiotics on the Structure of the Gut Microbial Community
3.4. Changes in Relative Abundance of the Gut Microbiome under the Chronic Exposure to Antibiotics
3.5. Changes in the Expression of Stress and Immune-Related Genes in Ricefish Chronically Exposed to Antibiotics
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Davies, J.; Davies, D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafhauser, B.H.; Kristofco, L.A.; de Oliveira, C.M.R.; Brooks, B.W. Global review and analysis of erythromycin in the environment: Occurrence, bioaccumulation and antibiotic resistance hazards. Environ. Pollut. 2018, 238, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Duan, Y.J.; Wang, S.P.; Wang, L.T.; Hou, Z.L.; Cui, Y.X.; Hou, J.; Das, R.; Mao, D.Q.; Luo, Y. Occurrence and distribution of clinical and veterinary antibiotics in the faeces of a Chinese population. J. Hazard. Mater. 2020, 383, 121129. [Google Scholar] [CrossRef]
- Cycon, M.; Mrozik, A.; Piotrowska-Seget, Z. Antibiotics in the Soil Environment-Degradation and Their Impact on Microbial Activity and Diversity. Front. Microbiol. 2019, 10, 338. [Google Scholar] [CrossRef]
- Yoon, J.B.; Hwang, S.; Baek, S.W.; Lee, S.; Bang, W.Y.; Moon, K.H. In vitro Edwardsiella piscicida CK108 Transcriptome Profiles with Subinhibitory Concentrations of Phenol and Formalin Reveal New Insights into Bacterial Pathogenesis Mechanisms. Microorganisms 2020, 8, 1068. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.N.; Farrag, H.A.; Aboulwafa, M.M.; Saleh, S.E. Effect of Subinhibitory Concentrations of Some Antibiotics and Low Doses of γ Radiation on the Cytotoxicity and Expression of Colibactin by an Uropathogenic Escherichia coli isolate. Curr. Microbiol. 2021, 78, 544–557. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.I.; Hughes, D. Microbiological effects of sublethal levels of antibiotics. Nat. Rev. Microbiol. 2014, 12, 465–478. [Google Scholar] [CrossRef]
- Lange, K.; Buerger, M.; Stallmach, A.; Bruns, T. Effects of Antibiotics on Gut Microbiota. Dig. Dis. 2016, 34, 260–268. [Google Scholar] [CrossRef]
- Yin, L.; Chen, B.; Xia, B.; Shi, X.; Qu, K. Polystyrene microplastics alter the behavior, energy reserve and nutritional composition of marine jacopever (Sebastes schlegelii). J. Hazard. Mater. 2018, 360, 97–105. [Google Scholar] [CrossRef]
- Chae, Y.; An, Y.J. Effects of micro-and nanoplastics on aquatic ecosystems: Current research trends and perspectives. Mar. Pollut. Bull. 2017, 124, 624–632. [Google Scholar] [CrossRef]
- Nichols, R.G.; Davenport, E.R. The relationship between the gut microbiome and host gene expression: A review. Hum. Genet. 2021, 140, 747–760. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Howitt, M.R.; Garrett, W.S. Exploring host-microbiota interactions in animal models and humans. Genes. Dev. 2013, 27, 701–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The effect of diet on the human gut microbiome: A metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 2009, 1, 6ra14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buford, T.W. (Dis)Trust your gut: The gut microbiome in age-related inflammation, health, and disease. Microbiome 2017, 5, 80. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.H.; Lin, G.; Fu, G.H.; Wan, Z.Y.; Lee, M.; Wang, L.; Liu, X.J.; Yue, G.H. The intestinal microbiome of fish under starvation. BMC Genom. 2014, 15, 266. [Google Scholar] [CrossRef] [Green Version]
- Clarke, G.; Sandhu, K.V.; Griffin, B.T.; Dinan, T.G.; Cryan, J.F.; Hyland, N.P. Gut Reactions: Breaking Down Xenobiotic-Microbiome Interactions. Pharmacol. Rev. 2019, 71, 198–224. [Google Scholar] [CrossRef]
- Francino, M.P. Antibiotics and the Human Gut Microbiome: Dysbioses and Accumulation of Resistances. Front. Microbiol. 2015, 6, 1543. [Google Scholar] [CrossRef] [Green Version]
- Weiss, G.A.; Hennet, T. Mechanisms and consequences of intestinal dysbiosis. Cell Mol. Life Sci. 2017, 74, 2959–2977. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.Y.; Chiang, C.Y.; Tsai, H.J. Zebrafish and Medaka: New model organisms for modern biomedical research. J. Biomed. Sci. 2016, 23, 19. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Chu, L.; Wojnarovits, L.; Takacs, E. Occurrence and fate of antibiotics, antibiotic resistant genes (ARGs) and antibiotic resistant bacteria (ARB) in municipal wastewater treatment plant: An overview. Sci. Total Environ. 2020, 744, 140997. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic. Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Collins, J.R.; Alvord, W.G.; Roayaei, J.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. The DAVID Gene Functional Classification Tool: A novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007, 8, R183. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, S.M.; Hargrave, B.T.; Haya, K. Antibiotic Use in Finfish Aquaculture: Modes of Action, Environmental Fate, and Microbial Resistance. In Environmental Effects of Marine Finfish Aquaculture; Hargrave, B.T., Ed.; Springer: Berlin/Heidelberg, Germany, 2005; pp. 341–357. [Google Scholar] [CrossRef]
- Chen, X.; Chen, H.; Liu, Q.; Ni, K.; Ding, R.; Wang, J.; Wang, C. High Plasticity of the Gut Microbiome and Muscle Metabolome of Chinese Mitten Crab (Eriocheir sinensis) in Diverse Environments. J. Microbiol. Biotechnol. 2021, 31, 240–249. [Google Scholar] [CrossRef]
- Aguilera, M.; Cerda-Cuellar, M.; Martinez, V. Antibiotic-induced dysbiosis alters host-bacterial interactions and leads to colonic sensory and motor changes in mice. Gut Microbes 2015, 6, 10–23. [Google Scholar] [CrossRef] [Green Version]
- Vallianou, N.; Dalamaga, M.; Stratigou, T.; Karampela, I.; Tsigalou, C. Do Antibiotics Cause Obesity Through Long-term Alterations in the Gut Microbiome? A Review of Current Evidence. Curr. Obes. Rep. 2021, 10, 244–262. [Google Scholar] [CrossRef]
- Kayani, M.U.R.; Yu, K.; Qiu, Y.; Shen, Y.; Gao, C.; Feng, R.; Zeng, X.; Wang, W.; Chen, L.; Su, H.L. Environmental concentrations of antibiotics alter the zebrafish gut microbiome structure and potential functions. Environ. Pollut. 2021, 278, 116760. [Google Scholar] [CrossRef]
- Petersen, C.; Round, J.L. Defining dysbiosis and its influence on host immunity and disease. Cell Microbiol. 2014, 16, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Larsson, D.G. Antibiotics in the environment. Ups. J. Med. Sci. 2014, 119, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Bu, Q.; Wang, B.; Huang, J.; Deng, S.; Yu, G. Pharmaceuticals and personal care products in the aquatic environment in China: A review. J. Hazard. Mater. 2013, 262, 189–211. [Google Scholar] [CrossRef] [PubMed]
- Shin, N.R.; Whon, T.W.; Bae, J.W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef]
- Kim, A.; Kim, N.; Roh, H.J.; Chun, W.K.; Ho, D.T.; Lee, Y.; Kim, D.H. Administration of antibiotics can cause dysbiosis in fish gut. Aquaculture 2019, 512, 734330. [Google Scholar] [CrossRef]
- Chung, T.H.; Yi, S.W.; Shin, G.W. Antibiotic resistance and repetitive-element PCR fingerprinting in Aeromonas veronii isolates. J. Fish Dis. 2017, 40, 821–829. [Google Scholar] [CrossRef]
- Lee, Y.; Choi, Y.; Lee, S.; Lee, H.; Kim, S.; Ha, J.; Lee, J.; Oh, H.; Kim, Y.; Yoon, Y. Occurrence of pathogenic Vibrio parahaemolyticus in seafood distribution channels and their antibiotic resistance profiles in S. Korea. Lett. Appl. Microbiol. 2019, 68, 128–133. [Google Scholar] [CrossRef]
- Shi, N.; Li, N.; Duan, X.; Niu, H. Interaction between the gut microbiome and mucosal immune system. Mil. Med. Res. 2017, 4, 14. [Google Scholar] [CrossRef]
- Wang, B.; Mao, C.; Feng, J.; Li, Y.; Hu, J.; Jiang, B.; Gu, Q.; Su, Y. A First Report of Aeromonas veronii Infection of the Sea Bass, Lateolabrax maculatus in China. Front. Vet. Sci. 2020, 7, 600587. [Google Scholar] [CrossRef]
- Zhang, Q.; Dong, X.; Chen, B.; Zhang, Y.; Zu, Y.; Li, W. Zebrafish as a useful model for zoonotic Vibrio parahaemolyticus pathogenicity in fish and human. Dev. Comp. Immunol. 2016, 55, 159–168. [Google Scholar] [CrossRef]
- Hoyt, M.A.; Stearns, T.; Botstein, D. Chromosome instability mutants of Saccharomyces cerevisiae that are defective in microtubule-mediated processes. Mol. Cell Biol. 1990, 10, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 2011, 117, 3720–3732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Guo, M.; Tang, X.; Xing, J.; Sheng, X.; Chi, H.; Zhan, W. Immune adjuvant effects of interferon-γ (IFN-γ) of flounder (Paralichthys olivaceus) against Edwardsiella tarda. Dev. Comp. Immunol. 2021, 123, 104159. [Google Scholar] [CrossRef] [PubMed]
- Goetz, F.W.; Planas, J.V.; MacKenzie, S. Tumor necrosis factors. Dev. Comp. Immunol. 2004, 28, 487–497. [Google Scholar] [CrossRef]
- Watts, C. The exogenous pathway for antigen presentation on major histocompatibility complex class II and CD1 molecules. Nat. Immunol. 2004, 5, 685–692. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Dijkstra, J.M. Major Histocompatibility Complex (MHC) Genes and Disease Resistance in Fish. Cells 2019, 8, 378. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, A.V.; Bartosch, B.; Isaguliants, M.G. Oxidative Stress in Infection and Consequent Disease. Oxid. Med. Cell Longev. 2017, 2017, 3496043. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Control | Ampicillin | p Value | Erythromycin | p Value |
|---|---|---|---|---|---|
| Chao1 | 60.33 ± 5.83 | 35.42 ± 7.66 | 0.001 | 43.52 ± 22.95 | 0.051 |
| Shannon | 2.42 ± 0.3 | 2.2 ± 0.03 | 0.087 | 1.78 ± 0.1 | 0.0001 |
| Inverse Simpson | 0.67 ± 0.08 | 0.68 ± 0.01 | 0.887 | 0.62 ± 0.02 | 0.096 |
| Good’s Coverage | 0.99 ± 0 | 0.99 ± 0 | 0.0001 | 0.99 ± 0 | 0.0001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoon, J.B.; Hwang, S.; Yang, J.H.; Lee, S.; Bang, W.Y.; Moon, K.H. Dynamics of the Gut Microbiome and Transcriptome in Korea Native Ricefish (Oryzias latipes) during Chronic Antibiotic Exposure. Genes 2022, 13, 1243. https://doi.org/10.3390/genes13071243
Yoon JB, Hwang S, Yang JH, Lee S, Bang WY, Moon KH. Dynamics of the Gut Microbiome and Transcriptome in Korea Native Ricefish (Oryzias latipes) during Chronic Antibiotic Exposure. Genes. 2022; 13(7):1243. https://doi.org/10.3390/genes13071243
Chicago/Turabian StyleYoon, Ju Bin, Sungmin Hwang, Jun Hyeok Yang, Seungki Lee, Woo Young Bang, and Ki Hwan Moon. 2022. "Dynamics of the Gut Microbiome and Transcriptome in Korea Native Ricefish (Oryzias latipes) during Chronic Antibiotic Exposure" Genes 13, no. 7: 1243. https://doi.org/10.3390/genes13071243