
Prediction of Alzheimer’s Disease by a Novel Image-Based Representation of Gene Expression




Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset and Data Preprocessing
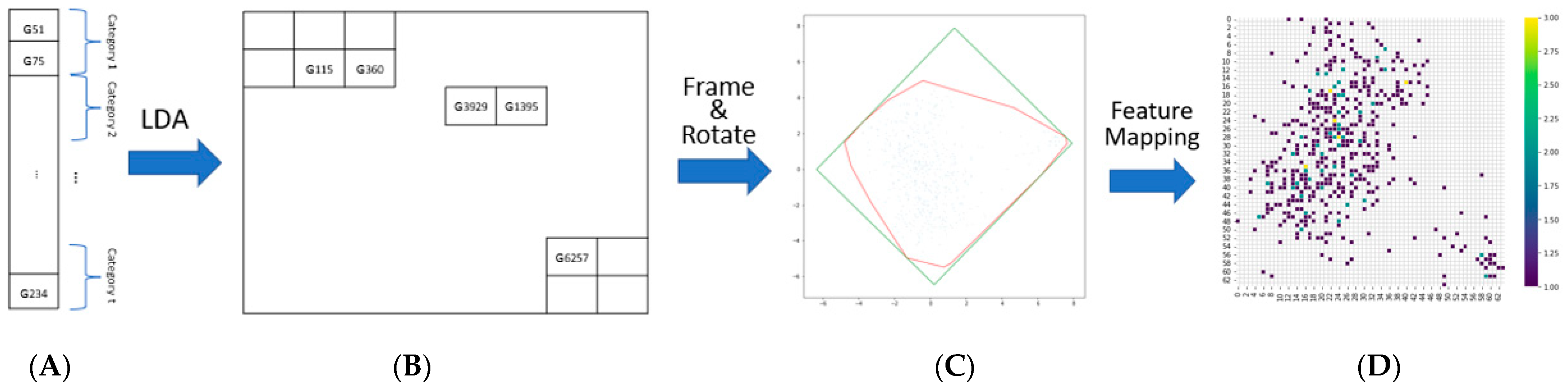
2.2. Image-Based Representation of mRNA Expression
2.3. Classification with Deep NN
3. Results
3.1. Image Representation Outputs
3.2. Pairwise Classification
3.3. Three-Class Classification
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mendez, M.F. Early-onset Alzheimer Disease and Its Variants. Continuum 2019, 25, 34–51. [Google Scholar] [CrossRef]
- Clark, R.; Hutton, M.; Fuldner, M.; Froelich, S.; Karran, E.; Talbot, C.; Crook, R.; Lendon, C.; Prihar, G.; He, C.; et al. The structure of the presenilin 1 (S182) gene and identification of six novel mutations in early onset AD families. Nat. Genet. 1995, 11, 219–222. [Google Scholar] [CrossRef] [PubMed]
- de la Vega, M.P.; Näslund, C.; Brundin, R.; Lannfelt, L.; Löwenmark, M.; Kilander, L.; Ingelsson, M.; Giedraitis, V. Mutation analysis of disease-causing genes in patients with early onset or familial forms of Alzheimer’s disease and frontotemporal dementia. BMC Genom. 2022, 23, 99. [Google Scholar]
- Wu, L.; Rosa-Neto, P.; Hsiung, G.-Y.R.; Sadovnick, A.D.; Masellis, M.; Black, S.E.; Jia, J.; Gauthier, S. Early-Onset Familial Alzheimer’s Disease (EOFAD). Can. J. Neurol. Sci. J. Can. Sci. Neurol. 2012, 39, 436–445. [Google Scholar] [CrossRef] [Green Version]
- Bagyinszky, E.; Youn, Y.C.; An, S.S.; Kim, S. The genetics of Alzheimer’s disease. Clin. Interv. Aging 2014, 9, 535–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koutsodendris, N.; Nelson, M.R.; Rao, A.; Huang, Y. Apolipoprotein E and Alzheimer’s disease: Findings, hypotheses, and potential mechanisms. Annu. Rev. Pathol. 2022, 17, 73–99. [Google Scholar] [CrossRef]
- Kamboh, M.I. Genomics and Functional Genomics of Alzheimer’s Disease. Neurotherapeutics 2022, 19, 152–172. [Google Scholar] [CrossRef]
- Dib, S.; Pahnke, J.; Gosselet, F. Role of ABCA7 in Human Health and in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 4603. [Google Scholar] [CrossRef]
- Khani, M.; Gibbons, E.; Bras, J.; Guerreiro, R. Challenge accepted: Uncovering the role of rare genetic variants in Alzheimer’s disease. Mol. Neurodegener. 2022, 17, 3. [Google Scholar] [CrossRef]
- Espinosa-Fernández, V.; Mañas-Ojeda, A.; Pacheco-Herrero, M.; Castro-Salazar, E.; Ros-Bernal, F.; Sánchez-Pérez, A.M. Early intervention with ABA prevents neuroinflammation and memory impairment in a triple transgenic mice model of Alzheimer´s disease. Behav. Brain Res. 2019, 374, 112106. [Google Scholar] [CrossRef] [PubMed]
- Ngandu, T.; Lehtisalo, J.; Solomon, A.; Levälahti, E.; Ahtiluoto, S.; Antikainen, R.; Bäckman, L.; Hänninen, T.; Jula, A.; Laatikainen, T.; et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): A randomised controlled trial. Lancet 2015, 385, 2255–2263. [Google Scholar] [CrossRef]
- Iso-Markku, P.; Kujala, U.M.; Knittle, K.; Polet, J.; Vuoksimaa, E.; Waller, K. Physical activity as a protective factor for dementia and Alzheimer’s disease: Systematic review, meta-analysis and quality assessment of cohort and case-control studies. Br. J. Sports Med. 2022, 56, 701–709. [Google Scholar] [CrossRef]
- Kumar, M.R.; Azizi, N.F.; Yeap, S.K.; Abdullah, J.O.; Khalid, M.; Omar, A.R.; Osman, M.A.; Leow, A.T.C.; Mortadza, S.A.S.; Alitheen, N.B. Clinical and Preclinical Studies of Fermented Foods and Their Effects on Alzheimer’s Disease. Antioxidants 2022, 11, 883. [Google Scholar] [CrossRef] [PubMed]
- Eid, A.; Mhatre, I.; Richardson, J.R. Gene-environment interactions in Alzheimer’s disease: A potential path to precision medicine. Pharmacol. Ther. 2019, 199, 173–187. [Google Scholar] [CrossRef]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-Analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escott-Price, V.; Bellenguez, C.; Wang, L.-S.; Choi, S.-H.; Harold, D.; Jones, L.; Holmans, P.; Gerrish, A.; Vedernikov, A.; Richards, A.; et al. Gene-Wide Analysis Detects Two New Susceptibility Genes for Alzheimer’s Disease. PLoS ONE 2014, 9, e94661. [Google Scholar] [CrossRef] [Green Version]
- Escott-Price, V.; Hardy, J. Genome-wide association studies for Alzheimer’s disease: Bigger is not always better. Brain Commun. 2022, 4, fcac125. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; Wang, R.; Zhang, Y.; Zhan, H. Prediction of Alzheimer’s Disease-Associated Genes by Integration of GWAS Summary Data and Expression Data. Front. Genet. 2019, 9, 653. [Google Scholar] [CrossRef]
- Farooq, A.; Anwar, S.; Awais, M.; Rehman, S. A deep CNN based multi-class classification of Alzheimer’s disease using MRI. In Proceedings of the IEEE International Conference on Imaging Systems and Techniques, Beijing, China, 18–20 October 2017. [Google Scholar] [CrossRef]
- Cui, R.; Liu, M. RNN-based longitudinal analysis for diagnosis of Alzheimer’s disease. Comput. Med. Imaging Graph. 2019, 73, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Lee, H. Prediction of Alzheimer’s disease using blood gene expression data. Sci. Rep. 2020, 10, 3485. [Google Scholar] [CrossRef] [PubMed]
- Mahendran, N.; Vincent, P.M.D.R.; Srinivasan, K.; Chang, C.-Y. Improving the Classification of Alzheimer’s Disease Using Hybrid Gene Selection Pipeline and Deep Learning. Front. Genet. 2021, 12, 784814. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, H.; Long, J.; Pan, G.; He, T.; Anichtchik, O.; Belshaw, R.; Albani, D.; Edison, P.; Green, E.K.; et al. Systematic Analysis and Biomarker Study for Alzheimer’s Disease. Sci. Rep. 2018, 8, 17394. [Google Scholar] [CrossRef]
- Yamashita, R.; Nishio, M.; Do, R.K.G.; Togashi, K. Convolutional neural networks: An overview and application in radiology. Insights Imaging 2018, 9, 611–629. [Google Scholar] [CrossRef] [Green Version]
- Brigato, L.; Iocchi, L. A close look at deep learning with small data. In Proceedings of the 25th International Conference on Pattern Recognition, Milan, Italy, 10–15 January 2020. [Google Scholar] [CrossRef]
- Sarraf, S.; Tofighi, G. Classification of Alzheimer’s Disease Using Fmri Data and Deep Learning Convolutional Neural Networks. arXiv 2016, arXiv:1603.08631. Available online: https://arxiv.org/abs/1603.08631 (accessed on 10 May 2022).
- Ji, H.; Liu, Z.; Yan, W.Q.; Klette, R. Early diagnosis of Alzheimer’s disease using deep learning. In Proceedings of the 2nd International Conference on Control and Computer Vision, Jeju, Korea, 15–18 June 2019. [Google Scholar] [CrossRef]
- Ramzan, F.; Khan, M.U.G.; Rehmat, A.; Iqbal, S.; Saba, T.; Rehman, A.; Mehmood, Z. A Deep Learning Approach for Automated Diagnosis and Multi-Class Classification of Alzheimer’s Disease Stages Using Resting-State fMRI and Residual Neural Networks. J. Med. Syst. 2020, 44, 37. [Google Scholar] [CrossRef] [PubMed]
- Bin Tufail, A.; Ma, Y.-K.; Zhang, Q.-N. Binary Classification of Alzheimer’s Disease Using sMRI Imaging Modality and Deep Learning. J. Digit. Imaging 2020, 33, 1073–1090. [Google Scholar] [CrossRef] [PubMed]
- Marzban, E.N.; Eldeib, A.M.; Yassine, I.A.; Kadah, Y.M.; Alzheimer’s Disease Neurodegenerative Initiative. Alzheimer’s disease diagnosis from diffusion tensor images using convolutional neural networks. PLoS ONE 2020, 15, e0230409. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Vans, E.; Shigemizu, D.; Boroevich, K.A.; Tsunoda, T. DeepInsight: A methodology to transform a non-image data to an image for convolution neural network architecture. Sci. Rep. 2019, 9, 11399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Maaten, L.; Hinton, G. Visualizing high-dimensional data using t-SNE. J. Mach. Learn. Res. 2008, 2579–2605. [Google Scholar]
- Amari, S.; Nagaoka, H. Methods of Information Geometry, Translations of Mathematical Monographs; American Mathematical Society: Providence, RI, USA, 2000; Volume 191. [Google Scholar]
- Sood, S.; Gallagher, I.J.; Lunnon, K.; Rullman, E.; Keohane, A.; Crossland, H.; Phillips, B.E.; Cederholm, T.; Jensen, T.; van Loon, L.J.; et al. A novel multi-tissue RNA diagnostic of healthy ageing relates to cognitive health status. Genome Biol. 2015, 16, 185. [Google Scholar] [CrossRef] [Green Version]
- Series GSE140829. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE140829 (accessed on 9 July 2022).
- Tibshirani, R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Ser. B Methodol. 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Krizhevsky, A.; Sutskever, I.; Hinton, G.E. Imagenet classification with deep convolutional neural networks. Adv. Neural Inf. Process. Syst. 2012, 25, 1106–1114. [Google Scholar] [CrossRef]
- Voyle, N.; Keohane, A.; Newhouse, S.; Lunnon, K.; Johnston, C.; Soininen, H.; Kloszewska, I.; Mecocci, P.; Tsolaki, M.; Vellas, B.; et al. A Pathway Based Classification Method for Analyzing Gene Expression for Alzheimer’s Disease Diagnosis. J. Alzheimer’s Dis. 2016, 49, 659–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Gawady, A.; Makhlouf, M.A.; Tawfik, B.S.; Nassar, H. Machine Learning Framework for the Prediction of Alzheimer’s Disease Using Gene Expression Data Based on Efficient Gene Selection. Symmetry 2022, 14, 491. [Google Scholar] [CrossRef]
- Guckiran, K.; Canturk, I.; Ozyilmaz, L. DNA microarray gene expression data classification using SVM, MLP, and RF with feature selection methods relief and LASSO. SDÜ Bilim. Enst. Derg. 2019, 23, 126–132. [Google Scholar]
- Wang, L.; Liu, Z.-P. Detecting Diagnostic Biomarkers of Alzheimer’s Disease by Integrating Gene Expression Data in Six Brain Regions. Front. Genet. 2019, 10, 157. [Google Scholar] [CrossRef]
- Park, C.; Ha, J.; Park, S. Prediction of Alzheimer’s disease based on deep neural network by integrating gene expression and DNA methylation dataset. Expert Syst. Appl. 2020, 140, 112873. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | GSE63060 | GSE63061 | GSE140829 | Combined Dataset |
|---|---|---|---|---|
| AD | 145 | 139 | 198 | 482 |
| MCI | 80 | 109 | 124 | 313 |
| CTL | 104 | 134 | 229 | 467 |
| Total | 329 | 382 | 551 | 1262 |
| Study | Method | Accuracy | AUC |
|---|---|---|---|
| El-Gawady et al. [39] | Multiple Feature Selection + SVM | 0.690 | 0.690 |
| Güçkıran et al. [40] | LASSO + SVM | 0.764 | 0.850 |
| Sharma et al. [31] | DeepInsight (tSNE + CNN) | 0.670 | 0.743 |
| Proposed Method | LDA-based imaging + CNN | 0.842 | 0.875 |
| Classes | Accuracy | AUC |
|---|---|---|
| AD vs. MCI | 0.704 | 0.664 |
| MCI vs. CTL | 0.698 | 0.619 |
| AD vs. (MCI and CTL) | 0.707 | 0.679 |
| (AD and MCI) vs. CTL | 0.773 | 0.742 |
| AD vs. CTL | 0.842 | 0.875 |
| AD | MCI | CTL | |
|---|---|---|---|
| AD | 0.71 | 0.19 | 0.10 |
| MCI | 0.08 | 0.76 | 0.15 |
| CTL | 0.36 | 0.38 | 0.25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalkan, H.; Akkaya, U.M.; Inal-Gültekin, G.; Sanchez-Perez, A.M. Prediction of Alzheimer’s Disease by a Novel Image-Based Representation of Gene Expression. Genes 2022, 13, 1406. https://doi.org/10.3390/genes13081406
Kalkan H, Akkaya UM, Inal-Gültekin G, Sanchez-Perez AM. Prediction of Alzheimer’s Disease by a Novel Image-Based Representation of Gene Expression. Genes. 2022; 13(8):1406. https://doi.org/10.3390/genes13081406
Chicago/Turabian StyleKalkan, Habil, Umit Murat Akkaya, Güldal Inal-Gültekin, and Ana Maria Sanchez-Perez. 2022. "Prediction of Alzheimer’s Disease by a Novel Image-Based Representation of Gene Expression" Genes 13, no. 8: 1406. https://doi.org/10.3390/genes13081406
APA StyleKalkan, H., Akkaya, U. M., Inal-Gültekin, G., & Sanchez-Perez, A. M. (2022). Prediction of Alzheimer’s Disease by a Novel Image-Based Representation of Gene Expression. Genes, 13(8), 1406. https://doi.org/10.3390/genes13081406