
An Analysis of Phenotype and Genotype in a Large Cohort of Chinese Children with Angelman Syndrome

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Recruitment
2.2. Study Design and Timing of Assessments
2.3. Statistical Analysis
3. Results
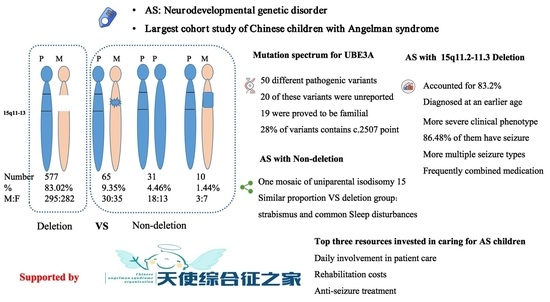
3.1. Patients and Genetic Studies
3.2. Mutation Spectrum for UBE3A Variants in Angelman Syndrome
3.3. Schematic Representation of Clinical Manifestations of Angelman Syndrome
3.4. Clinical Needs and Burden in Angelman Syndrome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Williams, C.A.; Frias, J.L. The Angelman (“happy puppet”) syndrome. Am. J. Med. Genet. 1982, 11, 453–460. [Google Scholar] [CrossRef]
- Petersen, M.B.; Brondum-Nielsen, K.; Hansen, L.K.; Wulff, K. Clinical, cytogenetic, and molecular diagnosis of Angelman syndrome: Estimated prevalence rate in a Danish county. Am. J. Med. Genet. 1995, 60, 261–262. [Google Scholar] [CrossRef]
- Mertz, L.G.; Christensen, R.; Vogel, I.; Hertz, J.M.; Nielsen, K.B.; Gronskov, K.; Ostergaard, J.R. Angelman syndrome in Denmark. birth incidence, genetic findings, and age at diagnosis. Am. J. Med. Genet. A 2013, 161, 2197–2203. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.K.; Glasson, E.J.; Bittles, A.H. A long-term population-based clinical and morbidity profile of Angelman syndrome in Western Australia: 1953–2003. Disabil. Rehabil. 2006, 28, 299–305. [Google Scholar] [CrossRef]
- Luk, H.M.; Lo, I.F. Angelman syndrome in Hong Kong Chinese: A 20 years’ experience. Eur. J. Med. Genet. 2016, 59, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.A.; Beaudet, A.L.; Clayton-Smith, J.; Knoll, J.H.; Kyllerman, M.; Laan, L.A.; Magenis, R.E.; Moncla, A.; Schinzel, A.A.; Summers, J.A.; et al. Angelman syndrome 2005: Updated consensus for diagnostic criteria. Am. J. Med. Genet. A 2006, 140, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Lev-Lehman, E.; Bressler, J.; Tsai, T.F.; Beaudet, A.L. Genetics of Angelman syndrome. Am. J. Hum. Genet. 1999, 65, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Maranga, C.; Fernandes, T.G.; Bekman, E.; da Rocha, S.T. Angelman syndrome: A journey through the brain. FEBS J. 2020, 287, 2154–2175. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.H.; Armstrong, D.; Albrecht, U.; Atkins, C.M.; Noebels, J.L.; Eichele, G.; Sweatt, J.D.; Beaudet, A.L. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron 1998, 21, 799–811. [Google Scholar] [CrossRef]
- Lopez, S.J.; Laufer, B.I.; Beitnere, U.; Berg, E.L.; Silverman, J.L.; O’Geen, H.; Segal, D.J.; LaSalle, J.M. Imprinting effects of UBE3A loss on synaptic gene networks and Wnt signaling pathways. Hum. Mol. Genet. 2019, 28, 3842–3852. [Google Scholar] [CrossRef] [PubMed]
- Buiting, K.; Clayton-Smith, J.; Driscoll, D.J.; Gillessen-Kaesbach, G.; Kanber, D.; Schwinger, E.; Williams, C.; Horsthemke, B. Clinical utility gene card for: Angelman Syndrome. Eur. J. Hum. Genet. 2015, 23, 3. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.L.; Qu, Y.J.; Jin, Y.W.; Wang, H.; Yang, Y.L.; Jiang, Y.W.; Yang, X.Y.; Zou, L.P.; Song, F. Molecular and clinical characterization of Angelman syndrome in Chinese patients. Clin. Genet. 2014, 85, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, S.; Harada, N.; Jinno, Y.; Hashimoto, K.; Imaizumi, K.; Kuroki, Y.; Fukushima, Y.; Sugimoto, T.; Renedo, M.; Wagstaff, J.; et al. Molecular and clinical study of 61 Angelman syndrome patients. Am. J. Med. Genet. 1994, 52, 158–163. [Google Scholar] [CrossRef]
- Wheeler, A.C.; Sacco, P.; Cabo, R. Unmet clinical needs and burden in Angelman syndrome: A review of the literature. Orphanet. J. Rare Dis. 2017, 12, 164. [Google Scholar] [CrossRef] [PubMed]
- Duis, J.; Nespeca, M.; Summers, J.; Bird, L.; Bindels-de Heus, K.; Valstar, M.J.; de Wit, M.Y.; Navis, C.; Ten Hooven-Radstaake, M.; van Iperen-Kolk, B.M.; et al. A multidisciplinary approach and consensus statement to establish standards of care for Angelman syndrome. Mol. Genet. Genomic Med. 2022, 10, e1843. [Google Scholar] [CrossRef] [PubMed]
- Bonello, D.; Camilleri, F.; Calleja-Agius, J. Angelman Syndrome: Identification and Management. Neonatal Netw. 2017, 36, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Wolter, J.M.; Mao, H.; Fragola, G.; Simon, J.M.; Krantz, J.L.; Bazick, H.O.; Oztemiz, B.; Stein, J.L.; Zylka, M.J. Cas9 gene therapy for Angelman syndrome traps Ube3a-ATS long non-coding RNA. Nature 2020, 587, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Markati, T.; Duis, J.; Servais, L. Therapies in preclinical and clinical development for Angelman syndrome. Expert Opin. Investig. Drugs 2021, 30, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Copping, N.A.; McTighe, S.M.; Fink, K.D.; Silverman, J.L. Emerging Gene and Small Molecule Therapies for the Neurodevelopmental Disorder Angelman Syndrome. Neurotherapeutics 2021, 18, 1535–1547. [Google Scholar] [CrossRef]
- Li, S.; Ma, Y.; Wang, T.; Jin, H.; Du, X.; Wang, Y. Epilepsy and Molecular Phenotype Affect the Neurodevelopment of Pediatric Angelman Syndrome Patients in China. Front. Psychiatry 2022, 13, 886028. [Google Scholar] [CrossRef] [PubMed]
- Camprubi, C.; Guitart, M.; Gabau, E.; Coll, M.D.; Villatoro, S.; Oltra, S.; Rosello, M.; Ferrer, I.; Monfort, S.; Orellana, C.; et al. Novel UBE3A mutations causing Angelman syndrome: Different parental origin for single nucleotide changes and multiple nucleotide deletions or insertions. Am. J. Med. Genet. A 2009, 149, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Ferrarini, A.; Xumerle, L.; Griggio, F.; Garonzi, M.; Cantaloni, C.; Centomo, C.; Vargas, S.M.; Descombes, P.; Marquis, J.; Collino, S.; et al. The Use of Non-Variant Sites to Improve the Clinical Assessment of Whole-Genome Sequence Data. PLoS ONE 2015, 10, e0132180. [Google Scholar] [CrossRef] [PubMed]
- Sadikovic, B.; Fernandes, P.; Zhang, V.W.; Ward, P.A.; Miloslavskaya, I.; Rhead, W.; Rosenbaum, R.; Gin, R.; Roa, B.; Fang, P. Mutation Update for UBE3A variants in Angelman syndrome. Hum. Mutat. 2014, 35, 1407–1417. [Google Scholar] [CrossRef] [PubMed]
- Lawson-Yuen, A.; Wu, B.L.; Lip, V.; Sahoo, T.; Kimonis, V. Atypical cases of Angelman syndrome. Am. J. Med. Genet. A 2006, 140, 2361–2364. [Google Scholar] [CrossRef] [PubMed]
- Aypar, U.; Hoppman, N.L.; Thorland, E.C.; Dawson, D.B. Patients with mosaic methylation patterns of the Prader-Willi/Angelman Syndrome critical region exhibit AS-like phenotypes with some PWS features. Mol. Cytogenet. 2016, 9, 26. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Miano, S.; Bruni, O.; Leuzzi, V.; Elia, M.; Verrillo, E.; Ferri, R. Sleep polygraphy in Angelman syndrome. Clin. Neurophysiol. 2004, 115, 938–945. [Google Scholar] [CrossRef]
- Thibert, R.L.; Conant, K.D.; Braun, E.K.; Bruno, P.; Said, R.R.; Nespeca, M.P.; Thiele, E.A. Epilepsy in Angelman syndrome: A questionnaire-based assessment of the natural history and current treatment options. Epilepsia 2009, 50, 2369–2376. [Google Scholar] [CrossRef]
- Shaaya, E.A.; Grocott, O.R.; Laing, O.; Thibert, R.L. Seizure treatment in Angelman syndrome: A case series from the Angelman Syndrome Clinic at Massachusetts General Hospital. Epilepsy Behav. 2016, 60, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Grocott, O.R.; Herrington, K.S.; Pfeifer, H.H.; Thiele, E.A.; Thibert, R.L. Low glycemic index treatment for seizure control in Angelman syndrome: A case series from the Center for Dietary Therapy of Epilepsy at the Massachusetts General Hospital. Epilepsy Behav. 2017, 68, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Wagstaff, J.; Knoll, J.H.; Fleming, J.; Kirkness, E.F.; Martin-Gallardo, A.; Greenberg, F.; Graham, J.M., Jr.; Menninger, J.; Ward, D.; Venter, J.C.; et al. Localization of the gene encoding the GABAA receptor β3 subunit to the Angelman/Prader-Willi region of human chromosome 15. Am. J. Hum. Genet. 1991, 49, 330–337. [Google Scholar]
- Quinn, E.D.; Rowland, C. Exploring Expressive Communication Skills in a Cross-Sectional Sample of Children and Young Adults with Angelman Syndrome. Am. J. Speech Lang Pathol. 2017, 26, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Sadhwani, A.; Wheeler, A.; Gwaltney, A.; Peters, S.U.; Barbieri-Welge, R.L.; Horowitz, L.T.; Noll, L.M.; Hundley, R.J.; Bird, L.M.; Tan, W.H. Developmental Skills of Individuals with Angelman Syndrome Assessed Using the Bayley-III. J. Autism. Dev. Disord. 2021, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Aghakhanyan, G.; Bonanni, P.; Randazzo, G.; Nappi, S.; Tessarotto, F.; De Martin, L.; Frijia, F.; De Marchi, D.; De Masi, F.; Kuppers, B.; et al. From Cortical and Subcortical Grey Matter Abnormalities to Neurobehavioral Phenotype of Angelman Syndrome: A Voxel-Based Morphometry Study. PLoS ONE 2016, 11, e0162817. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.M.; Jo, Y.; Shim, W.H.; Lee, J.S.; Ko, T.S.; Koo, J.H.; Yum, M.S. Disrupted Functional and Structural Connectivity in Angelman Syndrome. AJNR Am. J. Neuroradiol. 2020, 41, 889–897. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Total Number (n = 695) | Genotypes | ||||
|---|---|---|---|---|---|
| 15q11.2-q13del | UBE3A mutation | UPD15pat | Imprinting defect | Unclear (UPD15pat/ID) | |
| Number | 577 | 65 | 31 | 10 | 12 |
| % | 83.02% | 9.35% | 4.46% | 1.44% | 1.58% |
| M:F | 295:282 | 30:35 | 18:13 | 3:7 | 5:7 |
| Diagnosed age (months) | 24.19 ± 17.30 | 37.16 ± 38.89 | 30.12 ± 12.88 | 28.71 ± 13.06 | 38.31 ± 38.59 |
| Clinical Feature | Del (N = 577) /Per% | Non-Del (N = 118) /Per% | p | |||
|---|---|---|---|---|---|---|
| Epilepsy | 499 | 86.48% | 55 | 46.61% | <0.0001 (****) | |
| Sleep problems | 514 | 89.08% | 99 | 83.90% | NS | |
| Feeding problems | 480 | 83.19% | 84 | 71.19% | <0.01 (**) | |
| Speech impairment | 577 | 100% | 118 | 100% | NS | |
| No use of words | 463 | 80.24% | 60 | 50.85% | <0.0001 (****) | |
| Verbal (fewer than 2 words or word approximations) | 94 | 16.29% | 52 | 44.07% | <0.0001 (****) | |
| Short sentences | 5 | 0.87% | 6 | 5.08% | <0.001 (***) | |
| Facial features | Microcephaly | 295 | 51.13% | 48 | 40.68% | <0.05 (*) |
| Strabismus | 312 | 54.07% | 63 | 53.39% | NS | |
| Saprodontia | 185 | 32.06% | 48 | 40.68% | NS | |
| Widely spaced teeth | 310 | 53.73% | 59 | 50.00% | NS | |
| Light skin | 446 | 77.30% | 33 | 27.97% | <0.0001 (****) | |
| Obesity | 22 | 3.81% | 22 | 18.64% | <0.0001 (****) | |
| Scoliosis | 101 | 17.50% | 12 | 10.17% | <0.05 (*) | |
| Behavioral features | Hypermotoric | 544 | 94.28% | 103 | 87.29% | <0.01 (**) |
| Abnormal food-related behaviors | 462 | 80.07% | 80 | 67.80% | <0.001 (***) | |
| Fascination with crinkly items | 359 | 62.22% | 72 | 61.02% | NS | |
| Autotomy | 26 | 4.51% | 14 | 11.86% | <0.01 (**) | |
| Attraction to and fascination with water | 414 | 71.75% | 95 | 80.51% | NS | |
| Stereotyped behavior | 165 | 28.60% | 35 | 29.66% | NS | |
| Asphyxia due to foreign body | 53 | 9.19% | 14 | 11.86% | NS | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, X.; Wang, J.; Li, S.; Ma, Y.; Wang, T.; Wu, B.; Zhou, Y.; Yu, L.; Wang, Y. An Analysis of Phenotype and Genotype in a Large Cohort of Chinese Children with Angelman Syndrome. Genes 2022, 13, 1447. https://doi.org/10.3390/genes13081447
Du X, Wang J, Li S, Ma Y, Wang T, Wu B, Zhou Y, Yu L, Wang Y. An Analysis of Phenotype and Genotype in a Large Cohort of Chinese Children with Angelman Syndrome. Genes. 2022; 13(8):1447. https://doi.org/10.3390/genes13081447
Chicago/Turabian StyleDu, Xiaonan, Ji Wang, Shuang Li, Yu Ma, Tianqi Wang, Bingbing Wu, Yuanfeng Zhou, Lifei Yu, and Yi Wang. 2022. "An Analysis of Phenotype and Genotype in a Large Cohort of Chinese Children with Angelman Syndrome" Genes 13, no. 8: 1447. https://doi.org/10.3390/genes13081447
APA StyleDu, X., Wang, J., Li, S., Ma, Y., Wang, T., Wu, B., Zhou, Y., Yu, L., & Wang, Y. (2022). An Analysis of Phenotype and Genotype in a Large Cohort of Chinese Children with Angelman Syndrome. Genes, 13(8), 1447. https://doi.org/10.3390/genes13081447