
Characterizing the Mechanisms of Metalaxyl, Bronopol and Copper Sulfate against Saprolegnia parasitica Using Modern Transcriptomics


Abstract
:1. Introduction
2. Materials and Methods
2.1. Determination of MIC and MBC of Three Antimicrobial Agents
2.2. Total RNA Isolation
2.3. RNA Sequencing (RNA-seq) Library Construction and Sequencing
2.4. Sequencing, Data Processing and Quality Control
2.5. The Analysis of Differential Expression Genes (DEGs)
2.6. DEG-Enrichment Analysis
2.7. Protein Network Analysis of DEGs
2.8. RT qPCR
3. Results
3.1. The Antimicrobial Activity of Three Antimicrobials against S. parasitica
3.2. Illumina Sequencing and Quality Assessment
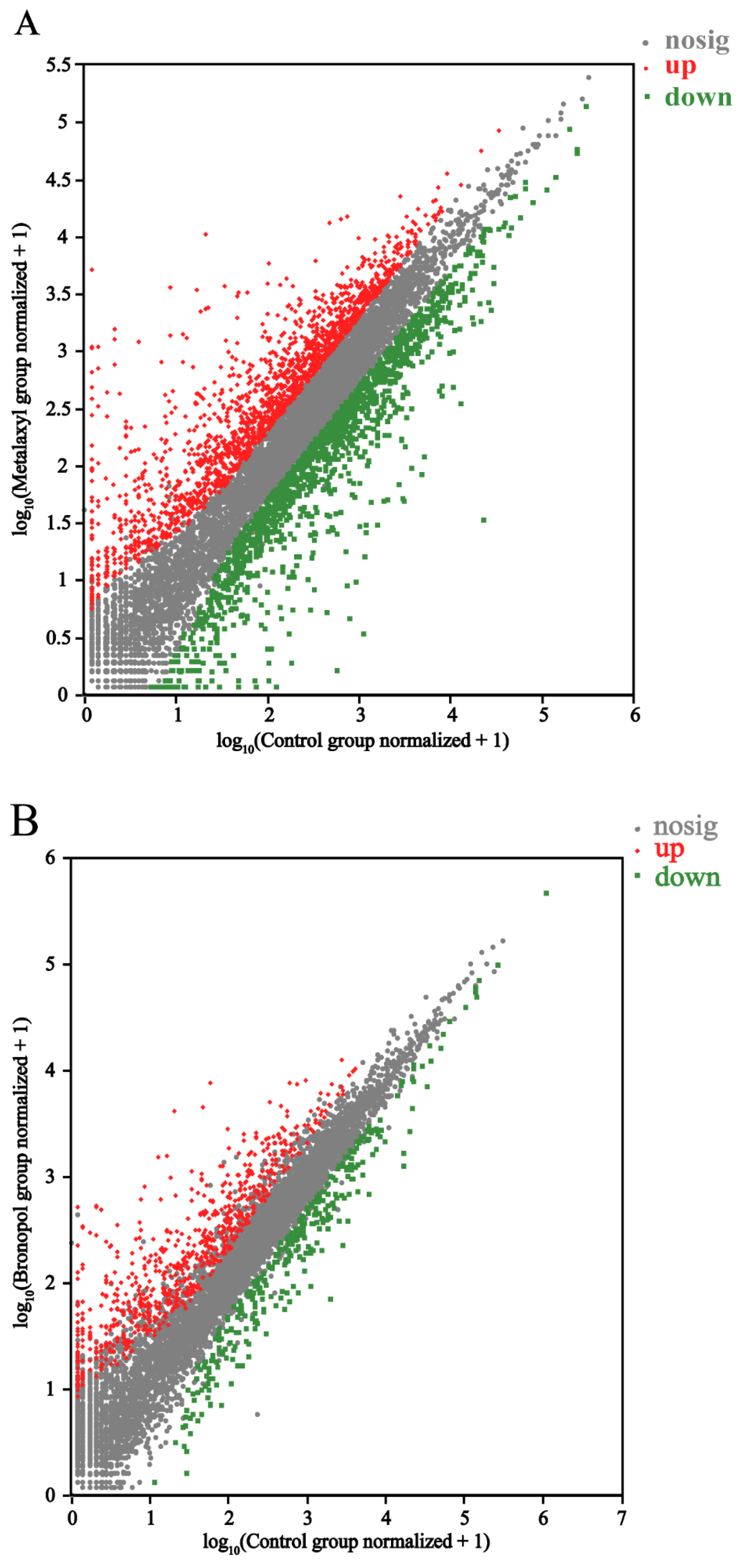
3.3. Effects of Three Antimicrobials on the Genome Expression of S. parasitica
3.4. Functional Annotation of DEGs
3.5. Enrichment Analysis of DEGs
3.6. Protein Network Analysis
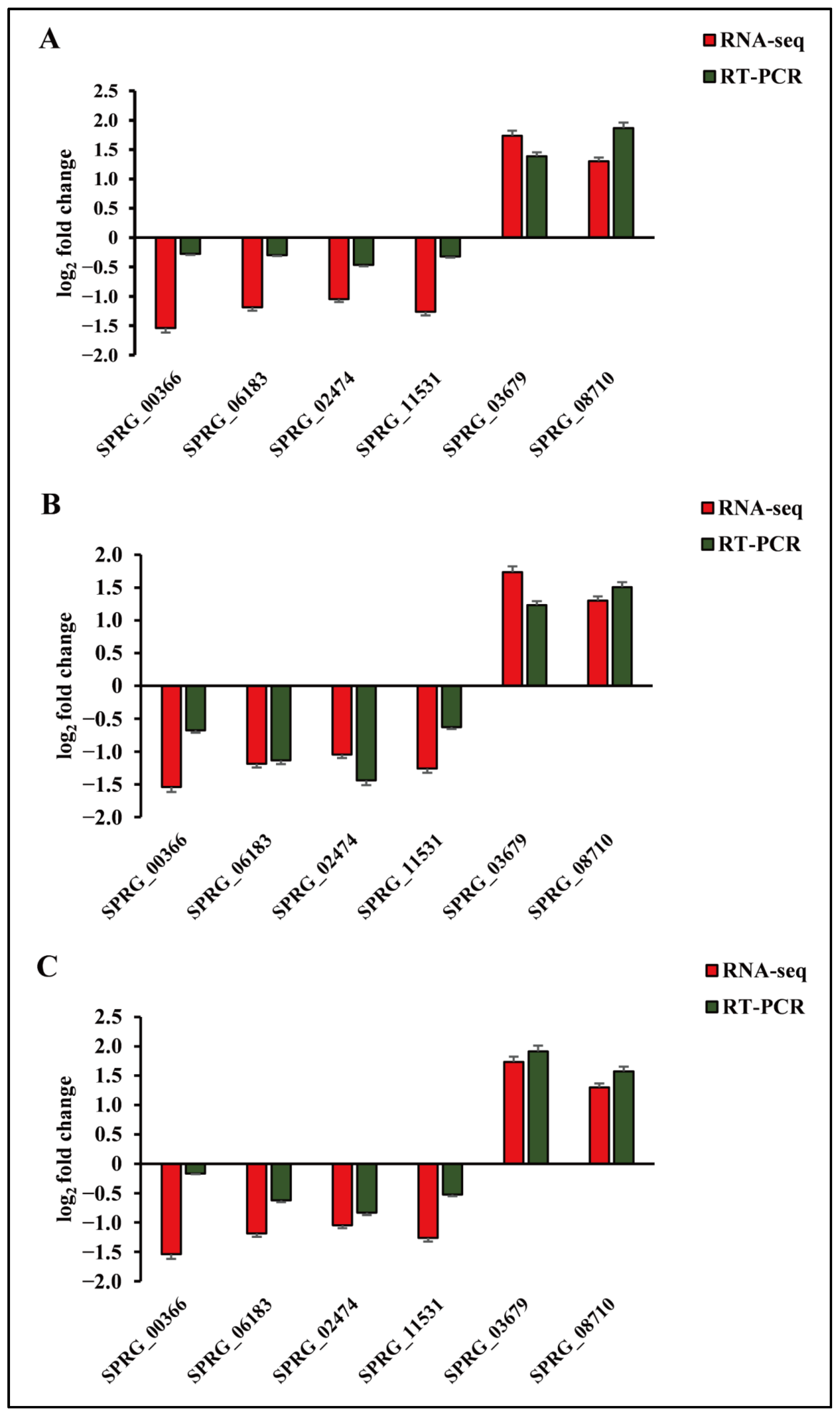
3.7. Verification of DEGs by qRT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Torto-Alalibo, T.; Tian, M.; Gajendran, K.; Waugh, M.E.; van West, P.; Kamoun, S. Expressed sequence tags from the oomycete fish pathogen Saprolegnia parasitica reveal putative virulence factors. BMC Microbiol. 2005, 5, 46. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.; Bruno, D. Fish Diseases and Disorders, Volume 3: Viral, Bacterial and Fungal Infections; CAB International Press: Wallingford, UK, 1999. [Google Scholar]
- van West, P. Saprolegnia parasitica, an oomycete pathogen with a fishy appetite: New challenges for an old problem. Mycologist 2006, 20, 99–104. [Google Scholar] [CrossRef]
- Bly, J.E.; Lawson, L.A.; Abdel-Aziz, E.S.; Clem, L.W. Channel Catfish, Ictalurus punctatus, Immunity to Saprolegnia sp. J. Appl. Aquac. 1994, 3, 35–50. [Google Scholar] [CrossRef]
- Sudova, E.; Machova, J.; Svobodova, Z.; Vesely, T. Negative effects of malachite green and possibilities of its replacement in the treatment of fish eggs and fish: A review. Vet. Med. 2007, 52, 527–539. [Google Scholar] [CrossRef]
- Zhang, R.; Zhou, Z. Effects of the Chiral Fungicides Metalaxyl and Metalaxyl-M on the Earthworm Eisenia fetida as Determined by H-1-NMR-Based Untargeted Metabolomics. Molecules 2019, 24, 1293. [Google Scholar] [CrossRef]
- van Jaarsveld, E.; Wingfield, M.J.; Drenth, A. Effect of metalaxyl resistance and cultivar resistance on control of Phytophthora nicotianae in tobacco. Plant Dis. 2002, 86, 362–366. [Google Scholar] [CrossRef]
- Grünwald, N.J.; Flier, W.G. The biology of Phytophthora infestans at its center of origin. Annu. Rev. Phytopathol. 2005, 43, 171–190. [Google Scholar] [CrossRef]
- Olea, A.F.; Espinoza, L.; Sedan, C.; Thomas, M.; Martínez, R.; Mellado, M.; Carrasco, H.; Díaz, K. Synthesis and In Vitro Growth Inhibition of 2-Allylphenol Derivatives Against Phythopthora cinnamomi Rands. Molecules 2019, 24, 4196. [Google Scholar] [CrossRef]
- Legin, G.Y. 2-Bromo-2-nitro-1,3-propanediol(Bronopol) and its derivatives: Synthesis, properties, and application (a review). Pharm. Chem. J. 1996, 30, 273–284. [Google Scholar] [CrossRef]
- Narenkumar, J.; Ramesh, N.; Rajasekar, A. Control of corrosive bacterial community by bronopol in industrial water system. 3 Biotech 2018, 8, 55. [Google Scholar] [CrossRef]
- Rousk, J.; Demoling, L.A.; Bahr, A.; Bååth, E. Examining the fungal and bacterial niche overlap using selective inhibitors in soil. FEMS Microbiol. Ecol. 2008, 63, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.J.; Anderson, R.C. Toxicity and metabolism of nitroalkanes and substituted nitroalkanes. J. Agric. Food Chem. 2013, 61, 763–779. [Google Scholar] [CrossRef] [PubMed]
- Jantrakajorn, S.; Wongtavatchai, J. Egg surface decontamination with bronopol increases larval survival of Nile tilapia, Oreochromis niloticus. Czech J. Anim. Sci. 2015, 60, 436–442. [Google Scholar] [CrossRef]
- Oono, H.; Hatai, K.; Miura, M.; Tuchida, N.; Kiryu, T. The use of bronopol to control fungal infection in rainbow trout eggs. Biocontrol Sci. 2007, 12, 55–57. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Shie, Y.Y.; Shiau, S.Y. Dietary copper requirements of juvenile grouper, Epinephelus malabaricus. Aquaculture 2008, 274, 161–165. [Google Scholar] [CrossRef]
- Straus, D.; Ledbetter, C.; Heikes, D. Inhibiting fungus on largemouth bass eggs with copper sulfate and its toxicity to fry and juveniles. J. World Aquac. Soc. 2019, 51, 214–223. [Google Scholar] [CrossRef]
- Mostafa, A.A.F.; Al-Askar, A.A.; Dawoud, T.M.; Ameen, F.; Yassin, M.T. In Vitro evaluation of antifungal activity of some agricultural fungicides against two saprolegnoid fungi infecting cultured fish. J. King Saud Univ. Sci. 2020, 32, 3091–3096. [Google Scholar] [CrossRef]
- Straus, D.L.; Farmer, B.D.; Ledbetter, C.K.; Beck, B.H.; Williams, R.S.; Clark, M.L.; Freeze, T.M. Use of Copper Sulfate to Control Egg Saprolegniasis at a Commercial Sunshine Bass Hatchery. N. Am. J. Aquac. 2016, 78, 243–250. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Jensen, L.J.; Kuhn, M.; Stark, M.; Chaffron, S.; Creevey, C.; Muller, J.; Doerks, T.; Julien, P.; Roth, A.; Simonovic, M.; et al. STRING 8—A global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009, 37, D412–D416. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-Protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.Z.; Liou, R.F. Selection of internal control genes for real-time quantitative RT-PCR assays in the oomycete plant pathogen Phytophthora parasitica. Fungal Genet. Biol. FG B 2006, 43, 430–438. [Google Scholar] [CrossRef] [PubMed]
- van West, P.; de Bruijn, I.; Minor, K.L.; Phillips, A.J.; Robertson, E.J.; Wawra, S.; Bain, J.; Anderson, V.L.; Secombes, C.J. The putative RxLR effector protein SpHtp1 from the fish pathogenic oomycete Saprolegnia parasitica is translocated into fish cells. FEMS Microbiol. Lett. 2010, 310, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.S.; Vestal, G.; Wille, K.; Patel, K.N.; Cheng, F.; Tipparaju, S.; Tousif, S.; Banday, M.M.; Xu, X.; Wilson, L.; et al. Differences in airway microbiome and metabolome of single lung transplant recipients. Respir. Res. 2020, 21, 104. [Google Scholar] [CrossRef]
- Hu, X.K.; Rao, S.S.; Tan, Y.J.; Yin, H.; Luo, M.J.; Wang, Z.X.; Zhou, J.H.; Hong, C.G.; Luo, Z.W.; Du, W.; et al. Fructose-coated Angstrom silver inhibits osteosarcoma growth and metastasis via promoting ROS-dependent apoptosis through the alteration of glucose metabolism by inhibiting PDK. Theranostics 2020, 10, 7710–7729. [Google Scholar] [CrossRef]
- Lapierre, F.M.; Schmid, J.; Ederer, B.; Ihling, N.; Büchs, J.; Huber, R. Revealing nutritional requirements of MICP-relevant Sporosarcina pasteurii DSM33 for growth improvement in chemically defined and complex media. Sci. Rep. 2020, 10, 22448. [Google Scholar] [CrossRef]
- Yang, Z.; Lu, R.; Dai, Z.; Yan, A.; Tang, Q.; Cheng, C.; Xu, Y.; Yang, W.; Su, J. Salt-Stress Response Mechanisms Using De Novo Transcriptome Sequencing of Salt-Tolerant and Sensitive Corchorus spp. Genotypes. Genes 2017, 8, 226. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Li, M.; Song, J.; Shi, Y.; Qin, X.; Gao, Z.; Lv, Y.; Du, G. The cardioprotective effects of the new crystal form of puerarin in isoproterenol-induced myocardial ischemia rats based on metabolomics. Sci. Rep. 2020, 10, 17787. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yu, H.; Chen, C.; Li, S.; Zhang, Z.; Xu, H.; Zhu, F.; Liu, J.; Spencer, P.S.; Dai, Z.; et al. Proteomic Profile of Mouse Brain Aging Contributions to Mitochondrial Dysfunction, DNA Oxidative Damage, Loss of Neurotrophic Factor, and Synaptic and Ribosomal Proteins. Oxidative Med. Cell. Longev. 2020, 2020, 5408452. [Google Scholar] [CrossRef] [PubMed]
- Hall, L.; Guo, C.; Tandy, S.; Broadhouse, K.; Dona, A.C.; Malle, E.; Bartels, E.D.; Christoffersen, C.; Grieve, S.M.; Figtree, G.; et al. Oral pre-treatment with thiocyanate (SCN(−)) protects against myocardial ischaemia-reperfusion injury in rats. Sci. Rep. 2021, 11, 12712. [Google Scholar] [CrossRef]
- Sung, M.H.; Tanizawa, K.; Tanaka, H.; Kuramitsu, S.; Kagamiyama, H.; Hirotsu, K.; Okamoto, A.; Higuchi, T.; Soda, K. Thermostable aspartate aminotransferase from a thermophilic Bacillus species. Gene cloning, sequence determination, and preliminary X-ray characterization. J. Biol. Chem. 1991, 266, 2567–2572. [Google Scholar] [CrossRef]
- Hadfield, A.; Kryger, G.; Ouyang, J.; Petsko, G.A.; Ringe, D.; Viola, R. Structure of aspartate-β-semialdehyde dehydrogenase from Escherichia coli, a key enzyme in the aspartate family of amino acid biosynthesis. J. Mol. Biol. 1999, 289, 991–1002. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Rezeng, C.; Wang, Y.; Li, J.; Zhang, L.; Chen, J.; Li, Z. Toxicological Risks of Renqingchangjue in Rats Evaluated by (1)H NMR-Based Serum and Urine Metabolomics Analysis. ACS Omega 2020, 5, 2169–2179. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, Y.; Kojima, H.; Tanaka, T. Mutational analysis of the feedback sites of lysine-sensitive aspartokinase of Escherichia coli. FEMS Microbiol. Lett. 1999, 173, 211–215. [Google Scholar] [CrossRef]
- Luo, H.Z.; Guan, Y.; Yang, R.; Qian, G.L.; Yang, X.H.; Wang, J.S.; Jia, A.Q. Growth inhibition and metabolomic analysis of Xanthomonas oryzae pv. oryzae treated with resveratrol. BMC Microbiol. 2020, 20, 117. [Google Scholar] [CrossRef]
- Abedin, M.J.; Wang, D.; McDonnell, M.A.; Lehmann, U.; Kelekar, A. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ. 2007, 14, 500–510. [Google Scholar] [CrossRef]
- Myneni, S.R.; Settem, R.P.; Sojar, H.T.; Malone, J.P.; Loimaranta, V.; Nakajima, T.; Sharma, A. Identification of a unique TLR2-interacting peptide motif in a microbial leucine-rich repeat protein. Biochem. Biophys. Res. Commun. 2012, 423, 577–582. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Qin, L.; Miao, S. Regulatory Mechanisms of L-Lactic Acid and Taste Substances in Chinese Acid Rice Soup (Rice-Acid) Fermented with a Lacticaseibacillus paracasei and Kluyveromyces marxianus. Front. Microbiol. 2021, 12, 594631. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.L.; Sun, Q.H.; Liu, G.R.; Guo, J.Y. Urinary Metabolomics Study of the Intervention Effect of Hypoglycemic Decoction on Type 2 Diabetes Mellitus Rats Model. Evid.-Based Complementary Altern. Med. 2019, 2019, 1394641. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, R.N. Structure, catalytic mechanism, and evolution of the glutathione transferases. Chem. Res. Toxicol. 1997, 10, 2–18. [Google Scholar] [CrossRef]
- Jakobsson, P.J.; Morgenstern, R.; Mancini, J.; Ford-Hutchinson, A.; Persson, B. Common structural features of MAPEG—A widespread superfamily of membrane associated proteins with highly divergent functions in eicosanoid and glutathione metabolism. Protein Sci. Publ. Protein Soc. 1999, 8, 689–692. [Google Scholar] [CrossRef]
- Herrick, J.; Sclavi, B. Ribonucleotide reductase and the regulation of DNA replication: An old story and an ancient heritage. Mol. Microbiol. 2007, 63, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Biou, V.; Dumas, R.; Cohen-Addad, C.; Douce, R.; Job, D.; Pebay-Peyroula, E. The crystal structure of plant acetohydroxy acid isomeroreductase complexed with NADPH, two magnesium ions and a herbicidal transition state analog determined at 1.65 A resolution. EMBO J. 1997, 16, 3405–3415. [Google Scholar] [CrossRef]
- Ouattara, B.; Duplessis, M.; Girard, C.L. Optimization and validation of a reversed-phase high performance liquid chromatography method for the measurement of bovine liver methylmalonyl-coenzyme a mutase activity. BMC Biochem. 2013, 14, 25. [Google Scholar] [CrossRef]
- Peters, H.L.; Pitt, J.J.; Wood, L.R.; Hamilton, N.J.; Sarsero, J.P.; Buck, N.E. Mouse models for methylmalonic aciduria. PLoS ONE 2012, 7, e40609. [Google Scholar] [CrossRef]
- Warren, B.E.; Lou, P.H.; Lucchinetti, E.; Zhang, L.; Clanachan, A.S.; Affolter, A.; Hersberger, M.; Zaugg, M.; Lemieux, H. Early mitochondrial dysfunction in glycolytic muscle, but not oxidative muscle, of the fructose-fed insulin-resistant rat. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E658–E667. [Google Scholar] [CrossRef]
- Zocher, G.; Vilstrup, J.; Heine, D.; Hallab, A.; Goralski, E.; Hertweck, C.; Stahl, M.; Schäberle, T.F.; Stehle, T. Structural basis of head to head polyketide fusion by CorB. Chem. Sci. 2015, 6, 6525–6536. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Fan, H.; Liang, R.; Zhang, R.; Zhang, J.; Zhu, J. Taraxacum officinale extract ameliorates dextran sodium sulphate-induced colitis by regulating fatty acid degradation and microbial dysbiosis. J. Cell. Mol. Med. 2019, 23, 8161–8172. [Google Scholar] [CrossRef] [PubMed]
- Beites, T.; Jansen, R.S.; Wang, R.; Jinich, A.; Rhee, K.Y.; Schnappinger, D.; Ehrt, S. Multiple acyl-CoA dehydrogenase deficiency kills Mycobacterium tuberculosis in vitro and during infection. Nat. Commun. 2021, 12, 6593. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, A.R.; Kakimoto, P.A.; Kowaltowski, A.J. Diet-sensitive sources of reactive oxygen species in liver mitochondria: Role of very long chain acyl-CoA dehydrogenases. PLoS ONE 2013, 8, e77088. [Google Scholar] [CrossRef]
- Bonds, A.C.; Yuan, T.; Werman, J.M.; Jang, J.; Lu, R.; Nesbitt, N.M.; Garcia-Diaz, M.; Sampson, N.S. Post-translational Succinylation of Mycobacterium tuberculosis Enoyl-CoA Hydratase EchA19 Slows Catalytic Hydration of Cholesterol Catabolite 3-Oxo-chol-4,22-diene-24-oyl-CoA. ACS Infect. Dis. 2020, 6, 2214–2224. [Google Scholar] [CrossRef] [PubMed]
- Xian, J.; Wang, N.; Zhao, P.; Zhang, Y.; Meng, J.; Ma, X.; Guo, X.; Wang, Z.; Bo, X. Molecular characterization and immune protection of the 3-hydroxyacyl-CoA dehydrogenase gene in Echinococcus granulosus. Parasites Vectors 2021, 14, 489. [Google Scholar] [CrossRef]
- Dietl, A.M.; Binder, U.; Bauer, I.; Shadkchan, Y.; Osherov, N.; Haas, H. Arginine Auxotrophy Affects Siderophore Biosynthesis and Attenuates Virulence of Aspergillus fumigatus. Genes 2020, 11, 423. [Google Scholar] [CrossRef]
- Haskins, N.; Panglao, M.; Qu, Q.; Majumdar, H.; Cabrera-Luque, J.; Morizono, H.; Tuchman, M.; Caldovic, L. Inversion of allosteric effect of arginine on N-acetylglutamate synthase, a molecular marker for evolution of tetrapods. BMC Biochem. 2008, 9, 24. [Google Scholar] [CrossRef]
- Oates, J.R.; McKell, M.C.; Moreno-Fernandez, M.E.; Damen, M.; Deepe, G.S., Jr.; Qualls, J.E.; Divanovic, S. Macrophage Function in the Pathogenesis of Non-alcoholic Fatty Liver Disease: The Mac Attack. Front. Immunol. 2019, 10, 2893. [Google Scholar] [CrossRef]
- Yang, X.X.; Wei, J.D.; Mu, J.K.; Liu, X.; Dong, J.C.; Zeng, L.X.; Gu, W.; Li, J.P.; Yu, J. Integrated metabolomic profiling for analysis of antilipidemic effects of Polygonatum kingianum extract on dyslipidemia in rats. World J. Gastroenterol. 2018, 24, 5505–5524. [Google Scholar] [CrossRef]
- Cui, Y.; Wang, Y.; Liu, M.; Qiu, L.; Xing, P.; Wang, X.; Ying, G.; Li, B. Determination of glucose deficiency-induced cell death by mitochondrial ATP generation-driven proton homeostasis. J. Mol. Cell Biol. 2017, 9, 395–408. [Google Scholar] [CrossRef] [Green Version]
- Murdaugh, L.S.; Wang, Z.; Del Priore, L.V.; Dillon, J.; Gaillard, E.R. Age-related accumulation of 3-nitrotyrosine and nitro-A2E in human Bruch’s membrane. Exp. Eye Res. 2010, 90, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Thao, M.T.; Karumanchi, D.K.; Yacout, S.M.; Gaillard, E.R. Nitrite ion modifies tyrosine and lysine residues of extracellular matrix proteins. Nitric Oxide Biol. Chem. 2018, 79, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Kusano, M.; Tabuchi, M.; Fukushima, A.; Funayama, K.; Diaz, C.; Kobayashi, M.; Hayashi, N.; Tsuchiya, Y.N.; Takahashi, H.; Kamata, A.; et al. Metabolomics data reveal a crucial role of cytosolic glutamine synthetase 1;1 in coordinating metabolic balance in rice. Plant J. Cell Mol. Biol. 2011, 66, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Mai, B.; Cai, T.; Luo, J.; Wu, W.; Liu, B.; Han, N.; Xing, F.; Deng, X. Optimization of a Binary Concrete Crack Self-Healing System Containing Bacteria and Oxygen. Materials 2017, 10, 116. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhao, J.; Zhao, Y.; Lu, X.; Zhou, Z.; Zhao, C.; Xu, G. Comprehensive investigation of tobacco leaves during natural early senescence via multi-platform metabolomics analyses. Sci. Rep. 2016, 6, 37976. [Google Scholar] [CrossRef]
- Klemke, F.; Beyer, G.; Sawade, L.; Saitov, A.; Korte, T.; Maldener, I.; Lockau, W.; Nürnberg, D.J.; Volkmer, T. All1371 is a polyphosphate-dependent glucokinase in Anabaena sp. PCC 7120. Microbiology 2014, 160, 2807–2819. [Google Scholar] [CrossRef]
- Cheng, M.L.; Ho, H.Y.; Liang, C.M.; Chou, Y.H.; Stern, A.; Lu, F.J.; Chiu, D.T. Cellular glucose-6-phosphate dehydrogenase (G6PD) status modulates the effects of nitric oxide (NO) on human foreskin fibroblasts. FEBS Lett. 2000, 475, 257–262. [Google Scholar] [CrossRef]
- Wang, W.; Jiang, W.; Liu, J.; Li, Y.; Gai, J.; Li, Y. Genome-wide characterization of the aldehyde dehydrogenase gene superfamily in soybean and its potential role in drought stress response. BMC Genomics. 2017, 18, 518. [Google Scholar] [CrossRef]
- Vasiliou, V.; Pappa, A.; Petersen, D.R. Role of aldehyde dehydrogenases in endogenous and xenobiotic metabolism. Chem.-Biol. Interact. 2000, 129, 1–19. [Google Scholar] [CrossRef]
- Jackson, B.; Brocker, C.; Thompson, D.C.; Black, W.; Vasiliou, K.; Nebert, D.W.; Vasiliou, V. Update on the aldehyde dehydrogenase gene (ALDH) superfamily. Hum. Genom. 2011, 5, 283–303. [Google Scholar] [CrossRef] [Green Version]
- Janati-Idrissi, R.; Junelles, A.M.; Petitdemange, H.; Gay, R. Regulation of coenzyme A transferase and acetoacetate decarboxylase activities in Clostridium acetobutylicum. Ann. L’Institut Pasteur Microbiol. 1988, 139, 683–688. [Google Scholar] [CrossRef]
- Jiang, R.H.; de Bruijn, I.; Haas, B.J.; Belmonte, R.; Löbach, L.; Christie, J.; van den Ackerveken, G.; Bottin, A.; Bulone, V.; Díaz-Moreno, S.M.; et al. Distinctive expansion of potential virulence genes in the genome of the oomycete fish pathogen Saprolegnia parasitica. PLoS Genet. 2013, 9, e1003272. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | MIC (mg/L) | MBC (mg/L) |
|---|---|---|
| Metalaxyl | 5 mg/L | 6 mg/L |
| Bronopol | 4 mg/L | 4 mg/L |
| Copper sulfate | 2 mg/L | 5 mg/L |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Wu, H.; Fei, S.; Zhang, J.; Hu, K. Characterizing the Mechanisms of Metalaxyl, Bronopol and Copper Sulfate against Saprolegnia parasitica Using Modern Transcriptomics. Genes 2022, 13, 1524. https://doi.org/10.3390/genes13091524
Wang Y, Wu H, Fei S, Zhang J, Hu K. Characterizing the Mechanisms of Metalaxyl, Bronopol and Copper Sulfate against Saprolegnia parasitica Using Modern Transcriptomics. Genes. 2022; 13(9):1524. https://doi.org/10.3390/genes13091524
Chicago/Turabian StyleWang, Yali, Haotian Wu, Siying Fei, Junzhe Zhang, and Kun Hu. 2022. "Characterizing the Mechanisms of Metalaxyl, Bronopol and Copper Sulfate against Saprolegnia parasitica Using Modern Transcriptomics" Genes 13, no. 9: 1524. https://doi.org/10.3390/genes13091524
APA StyleWang, Y., Wu, H., Fei, S., Zhang, J., & Hu, K. (2022). Characterizing the Mechanisms of Metalaxyl, Bronopol and Copper Sulfate against Saprolegnia parasitica Using Modern Transcriptomics. Genes, 13(9), 1524. https://doi.org/10.3390/genes13091524