Rational Approach to Finding Genes Encoding Molecular Biomarkers: Focus on Breast Cancer




Abstract
:1. Introduction
1.1. Breast Cancer
1.2. Traditional Approaches to Screening
1.3. Molecular Approaches to Clinical Diagnostics
1.4. Working Hypotheses
2. Materials and Methods
2.1. Selection of Known Markers for Use as Seeds in the Subsequent Analyses
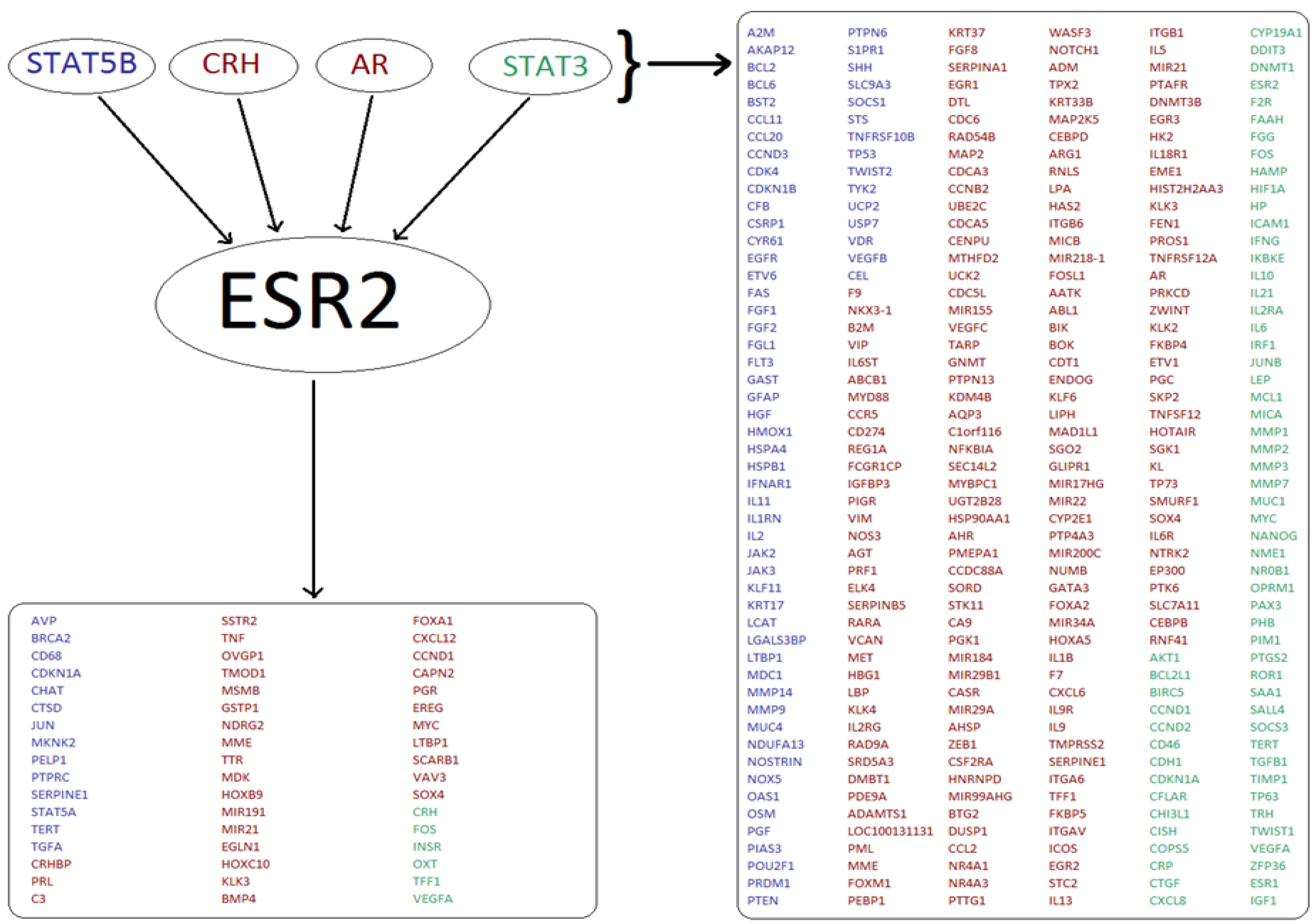
2.2. Transcriptional Networks
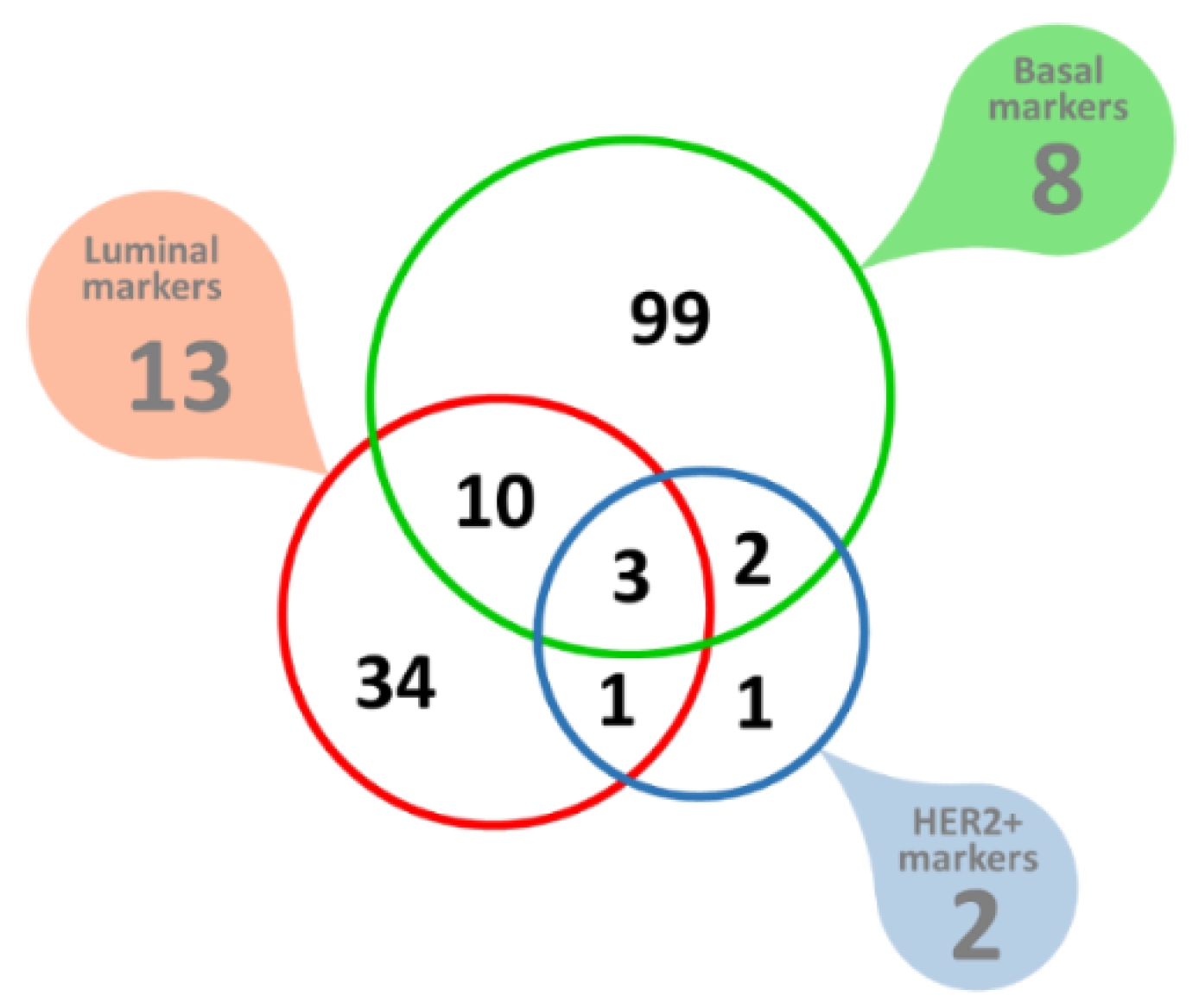
2.3. Biological Pathways
2.4. Gene Co-Expression and Protein–Protein Interactions
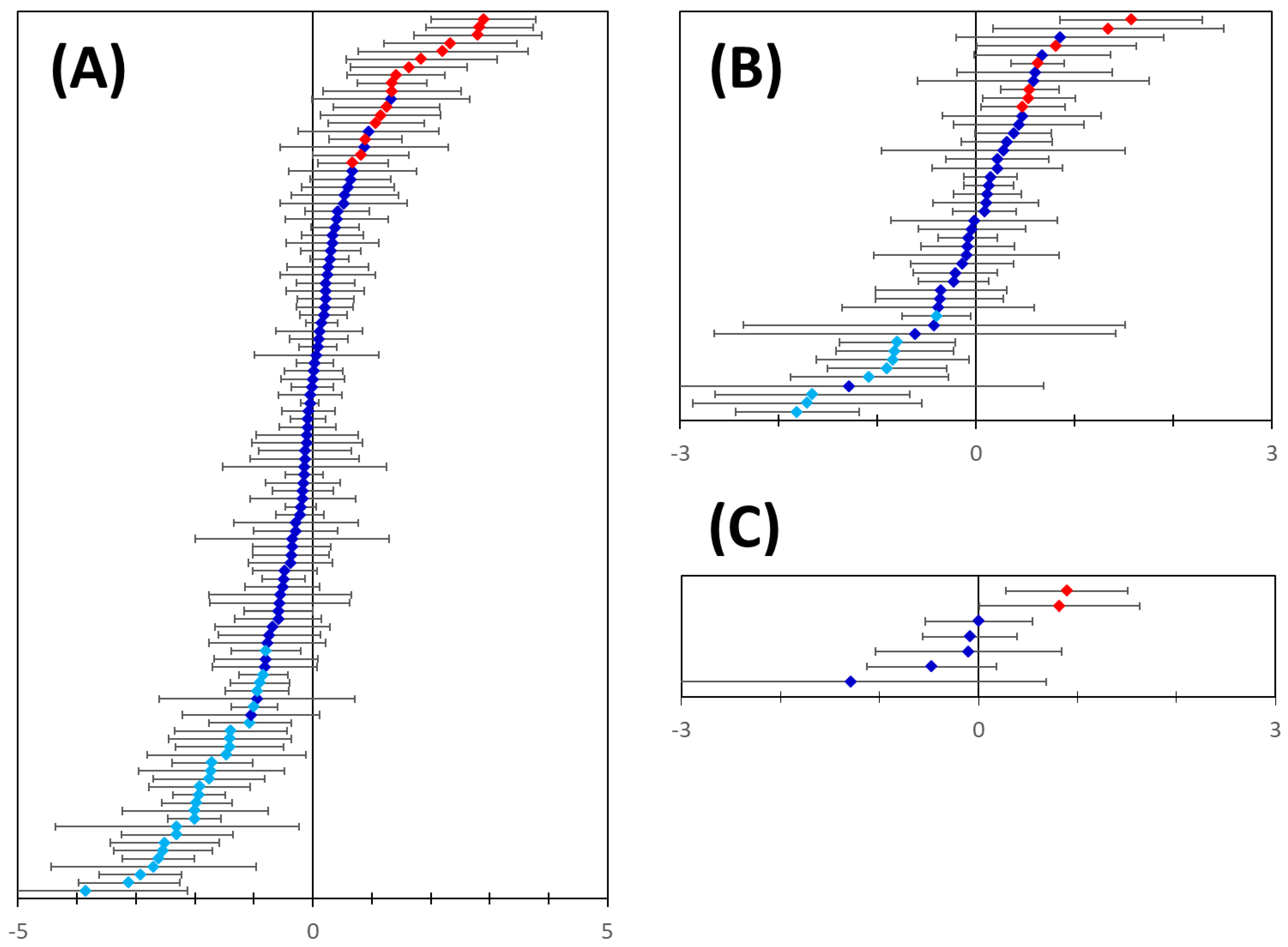
2.5. The Overall Prediction Strategy
2.6. Analysis of Protein Targeting and Further Marker Validation
3. Results
3.1. Analysis of Transcriptional Networks Yields Potentially Co-Regulated Genes
3.2. Analysis of Biological Pathways Identifies Potentially Functionally Related Genes
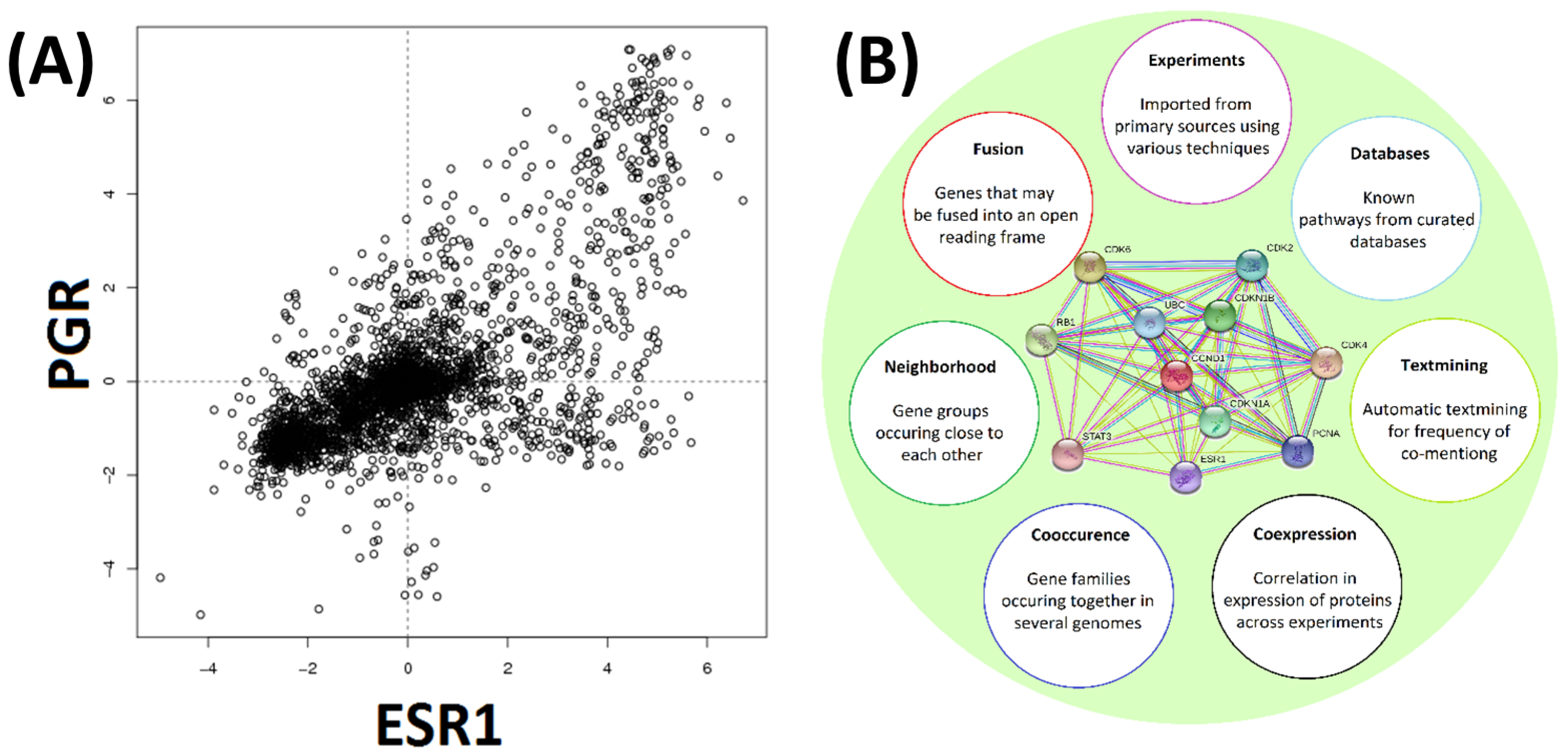
3.3. Exploring Gene Co-Expression and Protein Interaction Data
3.4. Exploring Gene Co-Expression and Protein Interaction Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Weigelt, B.; Geyer, F.C.; Reis-Filho, J.S. Histological Types of Breast Cancer: How Special are they? Mol. Oncol. 2010, 4, 192–208. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, C.; Ma, J.; Bryan, L.; Jemal, A. Breast Cancer Statistics, 2013. CA A Cancer J. Clin. 2013, 64, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Horlings, H.M.; Kreike, B.; Hayes, M.M.; Hauptmann, M.; Wessels, L.F.A.; de Jong, D.; Van de Vijver, M.J.; Van’t Veer, L.J.; Peterse, J.L. Refinement of Breast Cancer Classification by Molecular Characterization of Histological Special Types. J. Pathol. 2008, 216, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular Portraits of Human Breast Tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Naderi, A.; Teschendorff, A.E.; Barbosa-Morais, N.L.; Pinder, S.E.; Green, A.R.; Powe, D.G.; Robertson, J.F.R.; Aparicio, S.; Ellis, I.O.; Brenton, J.D.; et al. A Gene-Expression Signature to Predict Survival in Breast Cancer Across Independent Data Sets. Oncogene 2006, 26, 1507–1516. [Google Scholar] [CrossRef]
- Goldhirsch, A.; Wood, W.C.; Coates, A.S.; Gelber, R.D.; Thürlimann, B.; Senn, H. Strategies for Subtypes—dealing with the Diversity of Breast Cancer: Highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Ann. Oncol. 2011, 22, 1736–1747. [Google Scholar] [CrossRef]
- Prat, A.; Pineda, E.; Adamo, B.; Galván, P.; Fernández, A.; Gaba, L.; Díez, M.; Viladot, M.; Arance, A.; Muñoz, M. Clinical Implications of the Intrinsic Molecular Subtypes of Breast Cancer. Breast 2015, 24, S26–S35. [Google Scholar] [CrossRef]
- ISRCTNregistry. Available online: https://www.isrctn.com/ISRCTN33292440 (accessed on 25 March 2022).
- Long, H.; Brooks, J.M.; Harvie, M.; Maxwell, A.; French, D.P. Correction: How do Women Experience a False-Positive Test Result from Breast Screening? A Systematic Review and Thematic Synthesis of Qualitative Studies. Br. J. Cancer 2021, 125, 1031. [Google Scholar] [CrossRef]
- Mammograms. Available online: https://www.cancer.org/cancer/breast-cancer/screening-tests-and-early-detection/mammograms (accessed on 3 March 2022).
- Marmot, M.G.; Altman, D.G.; Cameron, D.A.; Dewar, J.A.; Thompson, S.G.; Wilcox, M. The Benefits and Harms of Breast Cancer Screening: An Independent Review. Br. J. Cancer 2013, 108, 2205–2240. [Google Scholar] [CrossRef]
- Ngan, T.T.; Nguyen, N.T.Q.; Van Minh, H.; Donnelly, M.; O’Neill, C. Effectiveness of Clinical Breast Examination as a ’Stand-Alone’ Screening Modality: An Overview of Systematic Reviews. BMC Cancer 2020, 20, 1070. [Google Scholar] [CrossRef]
- Chiarelli, A.M.; Majpruz, V.; Brown, P.; Thériault, M.; Shumak, R.; Mai, V. The Contribution of Clinical Breast Examination to the Accuracy of Breast Screening. J. Natl. Cancer Inst. 2009, 101, 1236–1243. [Google Scholar] [CrossRef] [PubMed]
- Wilson, N.; Ironside, A.; Diana, A.; Oikonomidou, O. Lobular Breast Cancer: A Review. Front. Oncol. 2021, 10, 591399. [Google Scholar] [CrossRef] [PubMed]
- Sood, R.; Rositch, A.F.; Shakoor, D.; Ambinder, E.; Pool, K.; Pollack, E.; Mollura, D.J.; Mullen, L.A.; Harvey, S.C. Ultrasound for Breast Cancer Detection Globally: A Systematic Review and Meta-Analysis. J. Glob. Oncol. 2019, 5, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Mann, R.M.; Kuhl, C.K.; Moy, L. Contrast-Enhanced MRI for Breast Cancer Screening. J. Magn. Reson. Imaging 2019, 50, 377–390. [Google Scholar] [CrossRef]
- Bick, U. Intensified Surveillance for Early Detection of Breast Cancer in High-Risk Patients. Breast Care 2015, 10, 13–20. [Google Scholar] [CrossRef]
- Salem, D.S.; Kamal, R.M.; Mansour, S.M.; Salah, L.A.; Wessam, R. Breast Imaging in the Young: The Role of Magnetic Resonance Imaging in Breast Cancer Screening, Diagnosis and Follow-Up. J. Thorac. Dis. 2013, 5, S9–S18. [Google Scholar]
- Lehman, C.D.; Lee, A.Y.; Lee, C.I. Imaging Management of Palpable Breast Abnormalities. Am. J. Roentgenol. 2014, 203, 1142–1153. [Google Scholar] [CrossRef]
- Heller, D.R.; Chiu, A.S.; Farrell, K.; Killelea, B.K.; Lannin, D.R. Why has Breast Cancer Screening Failed to Decrease the Incidence of De Novo Stage IV Disease? Cancers 2019, 11, 500. [Google Scholar] [CrossRef]
- Nelson, H.D.; Fu, R.; Cantor, A.; Pappas, M.; Daeges, M.; Humphrey, L. Effectiveness of Breast Cancer Screening: Systematic Review and Meta-Analysis to Update the 2009 U.S. Preventive Services Task Force Recommendation. Ann. Intern. Med. 2016, 164, 244–255. [Google Scholar] [CrossRef]
- D’Andrea, E.; Marzuillo, C.; De Vito, C.; Di Marco, M.; Pitini, E.; Vacchio, M.R.; Villari, P. Which BRCA Genetic Testing Programs are Ready for Implementation in Health Care? A Systematic Review of Economic Evaluations. Genet. Med. 2016, 18, 1171–1180. [Google Scholar] [CrossRef]
- Mukohara, T. PI3K Mutations in Breast Cancer: Prognostic and Therapeutic Implications. Breast Cancer Targets Ther. 2015, 7, 111–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Sáez, O.; Chic, N.; Pascual, T.; Adamo, B.; Vidal, M.; González-Farré, B.; Sanfeliu, E.; Schettini, F.; Conte, B.; Brasó-Maristany, F.; et al. Frequency and Spectrum of PIK3CA Somatic Mutations in Breast Cancer. Breast Cancer Res. 2020, 22, 45. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Cao, Y.; Liu, X.; Zeng, X.; Li, Y. Serum CA125 is a Predictive Marker for Breast Cancer Outcomes and Correlates with Molecular Subtypes. Oncotarget 2017, 8, 63963–63970. [Google Scholar] [CrossRef] [PubMed]
- Gaughran, G.; Aggarwal, N.; Shadbolt, B.; Stuart-Harris, R. The Utility of the Tumor Markers CA15.3, CEA, CA-125 and CA19.9 in Metastatic Breast Cancer. Breast Cancer Manag. 2020, 9, BMT50. [Google Scholar] [CrossRef]
- Colomer, R.; Ruibal, A.; Genollá, J.; Rubio, D.; Del Campo, J.M.; Bodi, R.; Salvador, L. Circulating CA 15-3 Levels in the Postsurgical Follow-Up of Breast Cancer Patients and in Non-Malignant Diseases. Breast Cancer Res. Treat. 1989, 13, 123–133. [Google Scholar] [CrossRef]
- Duffy, M.J.; Harbeck, N.; Nap, M.; Molina, R.; Nicolini, A.; Senkus, E.; Cardoso, F. Clinical use of Biomarkers in Breast Cancer: Updated Guidelines from the European Group on Tumor Markers (EGTM). Eur. J. Cancer 2017, 75, 284–298. [Google Scholar] [CrossRef]
- Malone, E.R.; Oliva, M.; Sabatini, P.J.B.; Stockley, T.L.; Siu, L.L. Molecular Profiling for Precision Cancer Therapies. Genome Med. 2020, 12, 8. [Google Scholar] [CrossRef]
- Prat Aparicio, A. Comprehensive Molecular Portraits of Human Breast Tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Asleh, K.; Negri, G.L.; Spencer Miko, S.E.; Colborne, S.; Hughes, C.S.; Wang, X.Q.; Gao, D.; Gilks, C.B.; Chia, S.K.L.; Nielsen, T.O.; et al. Proteomic Analysis of Archival Breast Cancer Clinical Specimens Identifies Biological Subtypes with Distinct Survival Outcomes. Nat. Commun. 2022, 13, 896. [Google Scholar] [CrossRef]
- Al-Wajeeh, A.S.; Salhimi, S.M.; Al-Mansoub, M.A.; Khalid, I.A.; Harvey, T.M.; Latiff, A.; Ismail, M.N. Comparative Proteomic Analysis of Different Stages of Breast Cancer Tissues using Ultra High Performance Liquid Chromatography Tandem Mass Spectrometer. PLoS ONE 2020, 15, e0227404. [Google Scholar] [CrossRef]
- Bouchal, P.; Schubert, O.T.; Faktor, J.; Capkova, L.; Imrichova, H.; Zoufalova, K.; Paralova, V.; Hrstka, R.; Liu, Y.; Ebhardt, H.A.; et al. Breast Cancer Classification Based on Proteotypes obtained by SWATH Mass Spectrometry. Cell Rep. 2019, 28, 832–843.e7. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Shim, H.; Shin, D.; Shim, J.E.; Ko, Y.; Shin, J.; Kim, H.; Cho, A.; Kim, E.; Lee, T.; et al. TRRUST: A Reference Database of Human Transcriptional Regulatory Interactions. Sci. Rep. 2015, 5, 11432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krull, M.; Pistor, S.; Voss, N.; Kel, A.; Reuter, I.; Kronenberg, D.; Michael, H.; Schwarzer, K.; Potapov, A.; Choi, C.; et al. TRANSPATH®: An Information Resource for Storing and Visualizing Signaling Pathways and their Pathological Aberrations. Nucleic Acids Res. 2006, 34, D546–D551. [Google Scholar] [CrossRef] [PubMed]
- Morandini, P. Rethinking Metabolic Control. Plant Sci. 2009, 176, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Giese, G.E.; Nanda, S.; Holdorf, A.D.; Walhout, A.J.M. Transcriptional Regulation of Metabolic Flux: A Caenorhabditis Elegans Perspective. Curr. Opin. Syst. Biol. 2019, 15, 12–18. [Google Scholar] [CrossRef]
- Moxley, J.F.; Jewett, M.C.; Antoniewicz, M.R.; Villas-Boas, S.G.; Alper, H.; Wheeler, R.T.; Tong, L.; Hinnebusch, A.G.; Ideker, T.; Nielsen, J.; et al. Linking High-Resolution Metabolic Flux Phenotypes and Transcriptional Regulation in Yeast Modulated by the Global Regulator Gcn4p. Proc. Natl. Acad. Sci. USA 2009, 106, 6477–6482. [Google Scholar] [CrossRef]
- Desvergne, B.; Michalik, L.; Wahli, W. Transcriptional Regulation of Metabolism. Physiol. Rev. 2006, 86, 465–514. [Google Scholar] [CrossRef]
- Van Dam, S.; Võsa, U.; van der Graaf, A.; Franke, L.; de Magalhães, J.P. Gene Co-Expression Analysis for Functional Classification and Gene-Disease Predictions. Brief. Bioinform. 2017, 19, 575–592. [Google Scholar] [CrossRef]
- TRANSPATH. Available online: http://genexplain.com/transpath/ (accessed on 25 March 2022).
- TRRUST. Available online: http://www.grnpedia.org/trrust/ (accessed on 25 March 2022).
- Botstein, D.; Cherry, J.M.; Ashburner, M.; Ball, C.A.; Blake, J.A.; Butler, H.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- GO. Available online: http://geneontology.org/ (accessed on 25 March 2022).
- NCBI. Available online: https://www.ncbi.nlm.nih.gov/protein (accessed on 25 March 2022).
- KEGG. Available online: http://www.genome.jp/kegg (accessed on 25 March 2022).
- Okamura, Y.; Aoki, Y.; Obayashi, T.; Tadaka, S.; Ito, S.; Narise, T.; Kinoshita, K. COXPRESdb in 2015: Coexpression Database for Animal Species by DNA-Microarray and RNAseq-Based Expression Data with Multiple Quality Assessment Systems. Nucleic Acids Res. 2015, 43, D82–D86. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-Protein Interaction Networks, Integrated Over the Tree of Life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- COXPRESdb. Available online: http://coxpresdb.jp/ (accessed on 25 March 2022).
- STRING. Available online: http://string-db.org/ (accessed on 25 March 2022).
- DAVID. Available online: https://david.ncifcrf.gov/conversion.jsp (accessed on 25 March 2022).
- UniProt. Available online: https://www.uniprot.org/ (accessed on 25 March 2022).
- Phobius. Available online: http://phobius.sbc.su.se/ (accessed on 25 March 2022).
- GSE124646. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE124646 (accessed on 25 March 2022).
- Reed, E.; Ferrari, E.; Soloviev, M. Quality Control of Gene Expression Data Allows Accurate Quantification of Differentially Expressed Biological Pathways. submitted.
- GPL96. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL96 (accessed on 3 October 2020).
- Califf, R.M. Biomarker Definitions and their Applications. Exp. Biol. Med. 2018, 243, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Burke, H.B. Predicting Clinical Outcomes using Molecular Biomarkers. Biomark. Cancer 2016, 8, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Kutomi, G.; Mizuguchi, T.; Satomi, F.; Maeda, H.; Shima, H.; Kimura, Y.; Hirata, K. Current Status of the Prognostic Molecular Biomarkers in Breast Cancer: A Systematic Review. Oncol. Lett. 2017, 13, 1491–1498. [Google Scholar] [CrossRef]
- Reis-Filho, J.S.; Pusztai, L. Gene Expression Profiling in Breast Cancer: Classification, Prognostication, and Prediction. Lancet 2011, 378, 1812–1823. [Google Scholar] [CrossRef]
- Russnes, H.G.; Navin, N.; Hicks, J.; Borresen-Dale, A. Insight into the Heterogeneity of Breast Cancer through Next-Generation Sequencing. J. Clin. Investig. 2011, 121, 3810–3818. [Google Scholar] [CrossRef]
- Michailidou, K.; Lindström, S.; Dennis, J.; Beesley, J.; Hui, S.; Kar, S.; Lemaçon, A.; Soucy, P.; Glubb, D.; Rostamianfar, A.; et al. Association analysis identifies 65 new breast cancer risk loci. Nature 2017, 551, 92–94. [Google Scholar] [CrossRef]
- Prosigna Breast Cancer Prognostic Gene Signature Assay. Available online: https://www.breastcancer.org/screening-testing/prosigna-assay (accessed on 30 March 2022).
- NanoString. Available online: https://nanostring.com/products/ncounter-assays-panels/oncology/ruo-pam50-codeset/ (accessed on 30 March 2022).
- Agendia: MammaPrint. Available online: https://agendia.com/mammaprint/ (accessed on 31 March 2022).
- Agendia: BluePrint. Available online: https://agendia.com/blueprint/ (accessed on 31 March 2022).
- Breast Cancer Index Test. Available online: https://www.breastcancer.org/screening-testing/breast-cancer-index-test (accessed on 31 March 2022).
- Breast Cancer Index. Available online: https://www.breastcancerindex.com (accessed on 31 March 2022).
- OncotypeIQ: Personalising Treatment Decisions. Available online: https://www.oncotypeiq.com/en-GB (accessed on 1 April 2022).
- Sotiriou, C.; Wirapati, P.; Loi, S.; Harris, A.; Fox, S.; Smeds, J.; Nordgren, H.; Farmer, P.; Praz, V.; Haibe-Kains, B.; et al. Gene Expression Profiling in Breast Cancer: Understanding the Molecular Basis of Histologic Grade to Improve Prognosis. J. Natl. Cancer Inst. 2006, 98, 262–272. [Google Scholar] [CrossRef]
- Dexur Pathway—Ventana Medical Systems. Available online: https://dexur.com/md/5046402/ (accessed on 3 April 2022).
- PATHVYSION HER-2 DNA PROBE KIT II. Available online: https://www.molecular.abbott/int/en/products/oncology/pathvysion-her-2-dna-probe-kit-II (accessed on 3 April 2022).
- Eccles, S.; Aboagye, E.; Ali, S.; Anderson, A.S.; Armes, J.; Berditchevski, F.; Blaydes, J.; Brennan, K.; Brown, N.; Bryant, H.; et al. Critical Research Gaps and Translational Priorities for the Successful Prevention and Treatment of Breast Cancer. Breast Cancer Res. 2013, 15, R92. [Google Scholar] [CrossRef]
- Cancer—Screening and Early Detection. Available online: https://www.euro.who.int/en/health-topics/noncommunicable-diseases/cancer/policy/screening-and-early-detection (accessed on 5 April 2022).
- Dibden, A.; Offman, J.; Duffy, S.W.; Gabe, R. Worldwide Review and Meta-Analysis of Cohort Studies Measuring the Effect of Mammography Screening Programmes on Incidence-Based Breast Cancer Mortality. Cancers 2020, 12, 976. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.; Garvican, L.; Tosteson, A.N.A.; Goodman, D.C.; Onega, T. Breast Cancer Screening in England and the United States: A Comparison of Provision and Utilisation. Int. J. Public Health 2015, 60, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Ahsen, M.E.; Chun, Y.; Grishin, A.; Grishina, G.; Stolovitzky, G.; Pandey, G.; Bunyavanich, S. NeTFactor, a framework for identifying transcriptional regulators of gene expression-based biomarkers. Sci. Rep. 2019, 9, 12970. [Google Scholar]
- Gambardella, G.; Moretti, M.N.; de Cegli, R.; Cardone, L.; Peron, A.; di Bernardo, D. Differential Network Analysis for the Identification of Condition-Specific Pathway Activity and Regulation. Bioinformatics 2013, 29, 1776–1785. [Google Scholar] [CrossRef] [Green Version]
- Debeljak, N.; Solár, P.; Sytkowski, A.J. Erythropoietin and cancer: The unintended consequences of anemia correction. Front Immunol. 2014, 11, 563. [Google Scholar] [CrossRef]
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef]
- Majumdar, S.; Diamandis, E.P. The promoter and the enhancer region of the KLK 3 (prostate specific antigen) gene is frequently mutated in breast tumours and in breast carcinoma cell lines. Br. J. Cancer 1999, 79, 1594–1602. [Google Scholar] [CrossRef] [Green Version]
- NCBI GEO. Available online: https://www.ncbi.nlm.nih.gov/geo/ (accessed on 12 February 2022).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | UniProt ID | Protein Name | HG-U133A 1 | B 2 | L 3 | H2 4 |
|---|---|---|---|---|---|---|
| BGN | P21810 | Biglycan | 201262_s_at | X | ||
| CEMIP | Q8WUJ3 | Cell migration-inducing and hyaluronan-binding protein | 212942_s_at | X | ||
| CXCL10 | P02778 | C-X-C motif chemokine 10 | 204533_at | X | ||
| CXCL8 | P10145 | Interleukin-8 | 211506_s_at | X | ||
| HPSE | Q9Y251 | Heparanase | 219403_s_at | X | ||
| INHBA | P08476 | Inhibin β A chain | 204926_at | X | ||
| MMP1 | P03956 | Interstitial collagenase | 204475_at | X | ||
| MMP11 | P24347 | Stromelysin-3 | 203878_s_at | X | ||
| MMP12 | P39900 | Macrophage metalloelastase | 204580_at | X | ||
| MMP13 | P45452 | Collagenase 3 | 205959_at | X | ||
| MMP9 | P14780 | Matrix metalloproteinase-9 | 203936_s_at | X | ||
| PLAU | P00749 | Urokinase-type plasminogen activator | 205479_s_at | X | ||
| PLAUR | Q03405 | Urokinase plasminogen activator surface receptor | 214866_at | X | ||
| FN1 | P02751 | Fibronectin | 214702_at | X | X | |
| VEGFA | P15692 | Vascular endothelial growth factor A | 210512_s_at | X | X | X |
| COL1A2 | P08123 | Collagen α-2(I) chain | 202404_s_at | X | X | |
| CTSD | P07339 | Cathepsin D | 200766_at | X | ||
| EDEM2 5 | Q9BV94 | ER degradation-enhancing α-mannosidase-like protein 2 | 218282_at | X | ||
| HSPA5 | P11021 | 78 kDa glucose-regulated protein | 211936_at | X | ||
| IFNG | P01579 | Interferon γ | 210354_at | X | ||
| IL18BP 5 | O95998 | Interleukin-18-binding protein | 219323_s_at | X |
| Gene | UniProt ID | Protein Name | HG-U133A 1 | B 2 | L 3 | H2 4 |
|---|---|---|---|---|---|---|
| ADAMTS1 | Q9UHI8 | Disintegrin and metalloproteinase with thrombospondin motifs 1 | 222162_s_at | X | ||
| BMP2 | P12643 | Bone morphogenetic protein 2 | 205289_at | X | ||
| BMP4 | P12644 | Bone morphogenetic protein 4 | 211518_s_at | X | ||
| CHRDL1 | Q9BU40 | Chordin-like protein 1 | 209763_at | X | ||
| CTGF | P29279 | Connective tissue growth factor | 209101_at | X | ||
| CYR61 | O00622 | Protein CYR61 | 201289_at | X | ||
| DCN | P07585 | Decorin | 209335_at | X | ||
| EDN1 | P05305 | Endothelin-1 | 218995_s_at | X | ||
| FBLN1 | P23142 | Fibulin-1 | 201787_at | X | ||
| FIGF | O43915 | Vascular endothelial growth factor D | 206742_at | X | ||
| FST | P19883 | Follistatin | 207345_at | X | ||
| IGF1 | P05019 | Insulin-like growth factor I | 209540_at | X | ||
| IGFBP3 | P17936 | Insulin-like growth factor-binding protein 3 | 210095_s_at | X | ||
| LAMC2 | Q13753 | Laminin subunit γ-2 | 202267_at | X | ||
| LEP | P41159 | Leptin | 207092_at | X | ||
| LPL | P06858 | Lipoprotein lipase | 203549_s_at | X | ||
| LTF | P02788 | Lactotransferrin | 202018_s_at | X | ||
| LUM | P51884 | Lumican | 201744_s_at | X | ||
| MFGE8 | Q08431 | Lactadherin | 210605_s_at | X | ||
| NID1 | P14543 | Nidogen-1 | 202007_at | X | ||
| OGN | P20774 | Mimecan | 218730_s_at | X | ||
| PDGFD | Q9GZP0 | Platelet-derived growth factor D | 219304_s_at | X | ||
| PENK | P01210 | Proenkephalin-A | 213791_at | X | ||
| PROS1 | P07225 | Vitamin K-dependent protein S | 207808_s_at | X | ||
| PTGDS | P41222 | Prostaglandin-H2 D-isomerase | 211663_x_at | X | ||
| PTGS2 | P35354 | Prostaglandin G/H synthase 2 | 204748_at | X | ||
| RELN | P78509 | Reelin | 205923_at | X | ||
| SOD3 | P08294 | Extracellular superoxide dismutase | 205236_x_at | X | ||
| PDGFA | P04085 | Platelet-derived growth factor subunit A | 205463_s_at | X | X | |
| ANG | P03950 | Angiogenin | 205141_at | X | ||
| C1S | P09871 | Complement C1s subcomponent | 208747_s_at | X | ||
| CD59 | P13987 | CD59 glycoprotein | 212463_at | X | ||
| KLK1 | P06870 | Kallikrein-1 | 216699_s_at | X | ||
| PTHLH | P12272 | Parathyroid hormone-related protein | 210355_at | X | ||
| SLPI | P03973 | Antileukoproteinase | 203021_at | X | ||
| SPARCL1 | Q14515 | SPARC-like protein 1 | 200795_at | X | ||
| TIMP3 | P35625 | Metalloproteinase inhibitor 3 | 201150_s_at | X |
| GEO Gene Ontology Biological Process 1 | Counts Observed 2 | Enrichment | p Value | ||
|---|---|---|---|---|---|
| Fold Difference over the Expected 3 | Rank 4 | ||||
| GO: 0030198 | extracellular matrix organization | 18 | 11.93 | 5 | 0 |
| GO: 0006508 | proteolysis | 14 | 6.50 | 10 | 6.6 × 10−16 |
| GO: 0007596 | blood coagulation | 12 | 5.24 | 11 | 1.3 × 10−10 |
| GO: 0022617 | extracellular matrix disassembly | 11 | 19.69 | 2 | 0 |
| GO: 0001525 | angiogenesis | 10 | 9.05 | 8 | 0 |
| GO: 0007275 | multicellular organismal development | 10 | 2.70 | 18 | 10−3 |
| GO: 0008284 | positive regulation of cell proliferation | 10 | 5.21 | 12 | 5.1 × 10−09 |
| GO: 0008285 | negative regulation of cell proliferation | 10 | 5.09 | 13 | 9.2 × 10−09 |
| GO: 0044267 | cellular protein metabolic process | 9 | 3.96 | 14 | 7.9 × 10−06 |
| GO: 0045944 | positive regulation of transcription | 9 | 2.43 | 20 | 5.7 × 10−3 |
| GO: 0030154 | cell differentiation | 8 | 3.05 | 17 | 8.6 × 10−4 |
| GO: 0030168 | platelet activation | 8 | 7.60 | 9 | 1.2 × 10−11 |
| GO: 0030335 | positive regulation of cell migration | 8 | 12.46 | 4 | 0 |
| GO: 0043066 | negative regulation of apoptotic process | 8 | 3.42 | 15 | 2.1 × 10−4 |
| GO: 0001666 | response to hypoxia | 8 | 9.46 | 7 | 7.1 × 10−15 |
| GO: 0007155 | cell adhesion | 8 | 3.14 | 16 | 6.3 × 10−4 |
| GO: 0030574 | collagen catabolic process | 7 | 20.78 | 1 | 0 |
| GO: 0001501 | skeletal system development | 7 | 10.08 | 6 | 3.7 × 10−14 |
| GO: 0002576 | platelet degranulation | 7 | 14.26 | 3 | 0 |
| GO: 0000122 | negative regulation of transcription | 7 | 2.63 | 19 | 7.6 × 10−3 |
| GEO Gene Ontology Cellular Components 1 | Counts Observed 2 | Enrichment | p Value | ||
|---|---|---|---|---|---|
| Fold Difference over the Expected 3 | Rank | ||||
| GO: 0005576 | extracellular region | 55 | 8.90 | 8 | 0 |
| GO: 0005615 | extracellular space | 36 | 8.41 | 9 | 0 |
| GO: 0070062 | extracellular vesicular exosome | 26 | 2.56 | 11 | 4.1 × 10−07 |
| GO: 0005578 | proteinaceous extracellular matrix | 22 | 19.92 | 3 | 0 |
| GO: 0031012 | extracellular matrix | 20 | 20.99 | 2 | 0 |
| GO: 0005604 | basement membrane | 8 | 17.21 | 6 | 0 |
| GO: 0009986 | cell surface | 8 | 3.71 | 10 | 6.4 × 10−05 |
| GO: 0005796 | Golgi lumen | 7 | 18.75 | 4 | 0 |
| GO: 0031093 | platelet α granule lumen | 6 | 22.31 | 1 | 0 |
| GO: 0043202 | lysosomal lumen | 6 | 17.28 | 5 | 0 |
| GO: 0005788 | endoplasmic reticulum lumen | 6 | 8.98 | 7 | 6.6 × 10−11 |
| GO: 0005789 | endoplasmic reticulum membrane | 6 | 2.23 | 12 | 4.3 × 10−2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schneider, N.; Reed, E.; Kamel, F.; Ferrari, E.; Soloviev, M. Rational Approach to Finding Genes Encoding Molecular Biomarkers: Focus on Breast Cancer. Genes 2022, 13, 1538. https://doi.org/10.3390/genes13091538
Schneider N, Reed E, Kamel F, Ferrari E, Soloviev M. Rational Approach to Finding Genes Encoding Molecular Biomarkers: Focus on Breast Cancer. Genes. 2022; 13(9):1538. https://doi.org/10.3390/genes13091538
Chicago/Turabian StyleSchneider, Nathalie, Ellen Reed, Faddy Kamel, Enrico Ferrari, and Mikhail Soloviev. 2022. "Rational Approach to Finding Genes Encoding Molecular Biomarkers: Focus on Breast Cancer" Genes 13, no. 9: 1538. https://doi.org/10.3390/genes13091538
APA StyleSchneider, N., Reed, E., Kamel, F., Ferrari, E., & Soloviev, M. (2022). Rational Approach to Finding Genes Encoding Molecular Biomarkers: Focus on Breast Cancer. Genes, 13(9), 1538. https://doi.org/10.3390/genes13091538