
Comparative Mitogenomics of Two Sympatric Catfishes of Exostoma (Siluriformes: Sisoridae) from the Lower Yarlung Tsangpo River and Its Application for Phylogenetic Consideration



Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling and DNA Extraction
2.2. Next-Generation Sequencing and Libraries’ Construction
2.3. Mitogenome Assembly, Annotation, and Analysis
2.4. Phylogenetic Analysis
2.5. Divergence Time Estimation
2.6. Selection Pressure Analysis
3. Results and Discussion
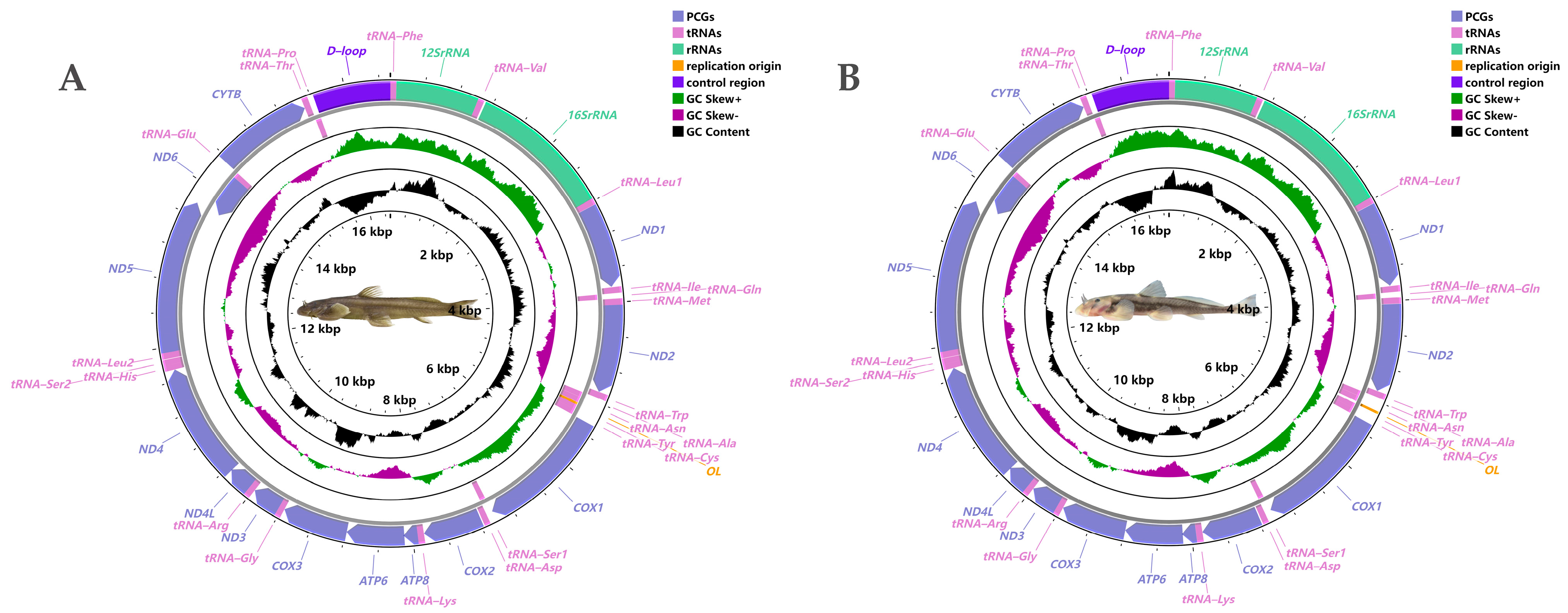
3.1. Mitogenome Architecture and Composition
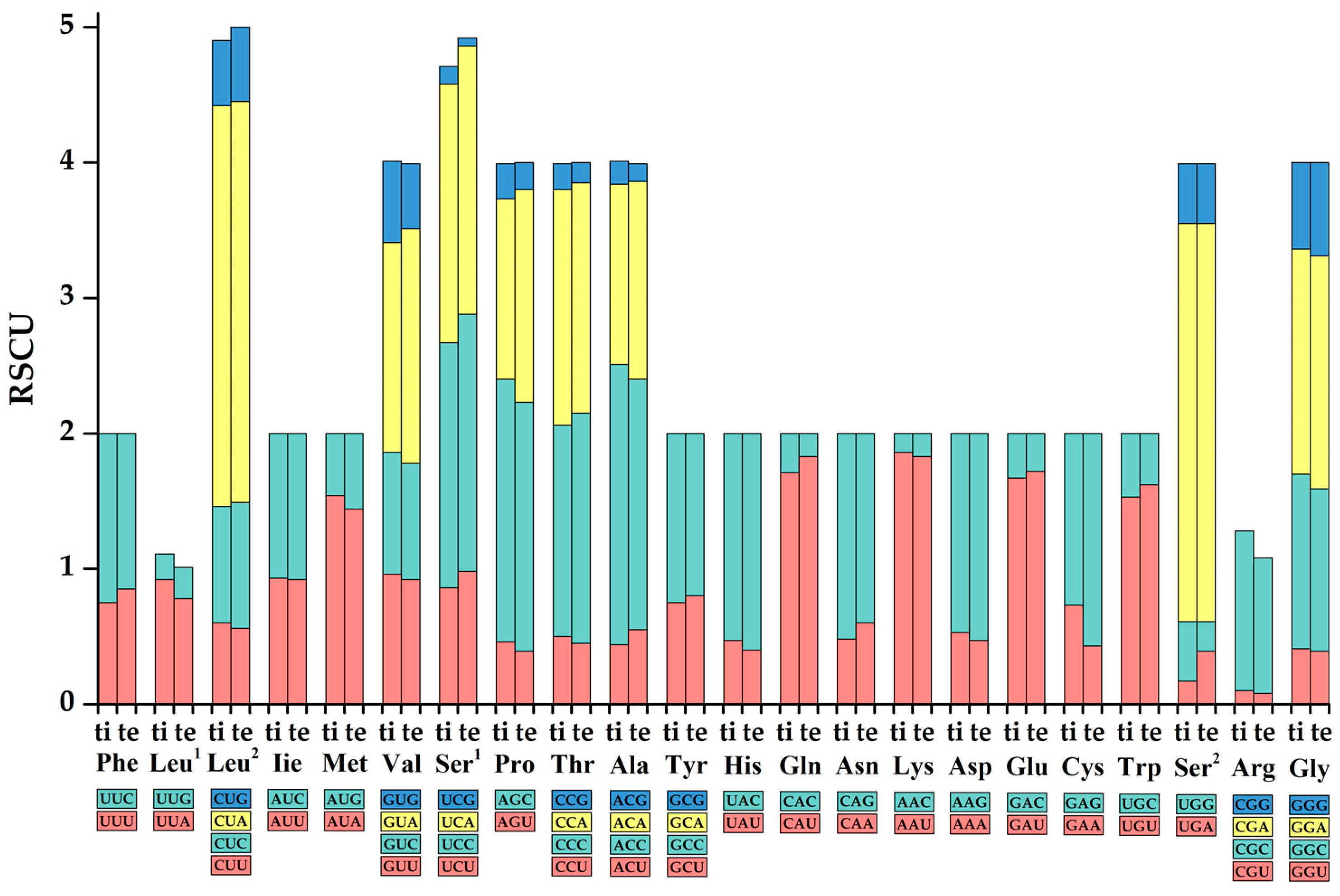
3.2. Protein-Coding Genes and Codon Usage
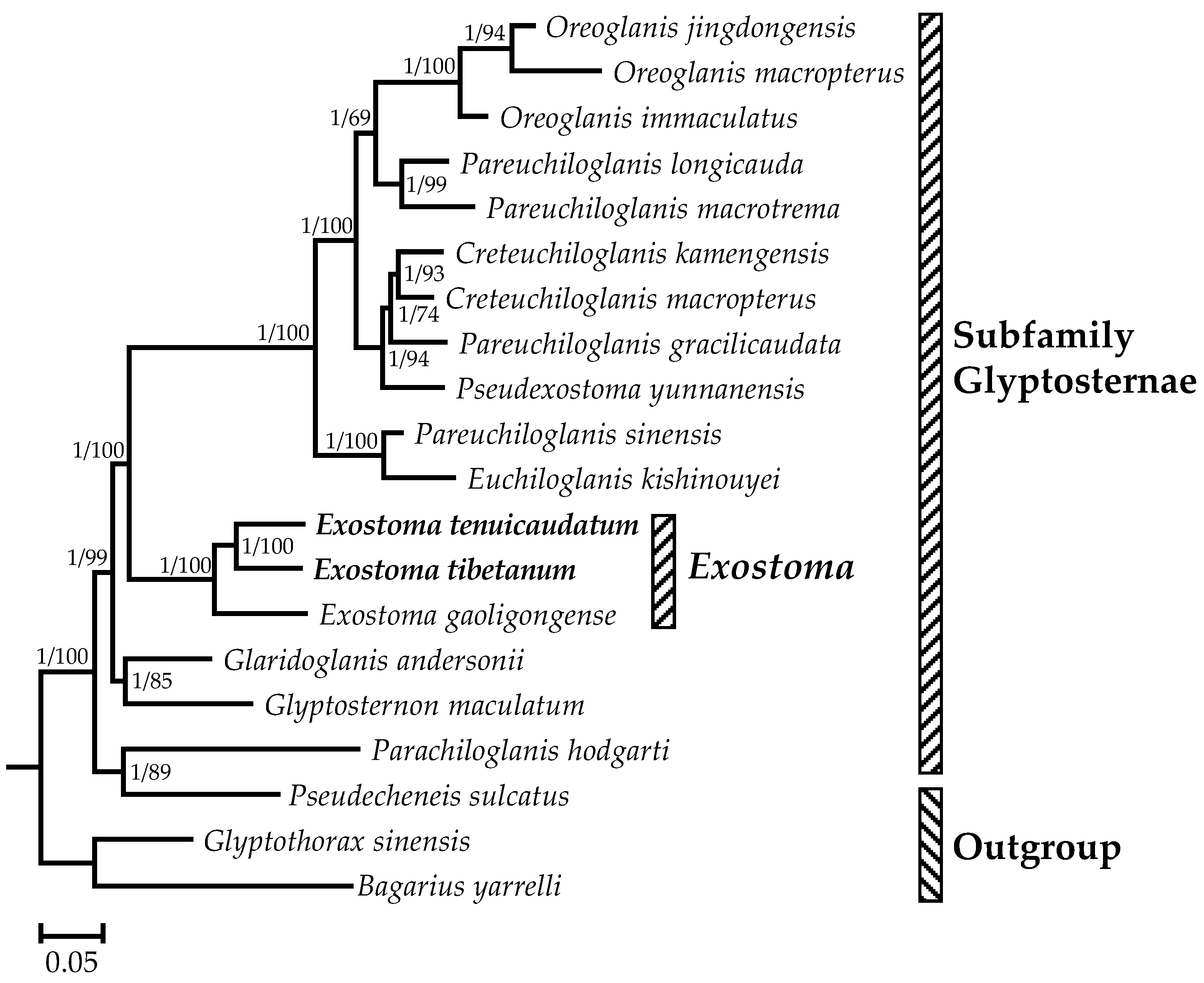
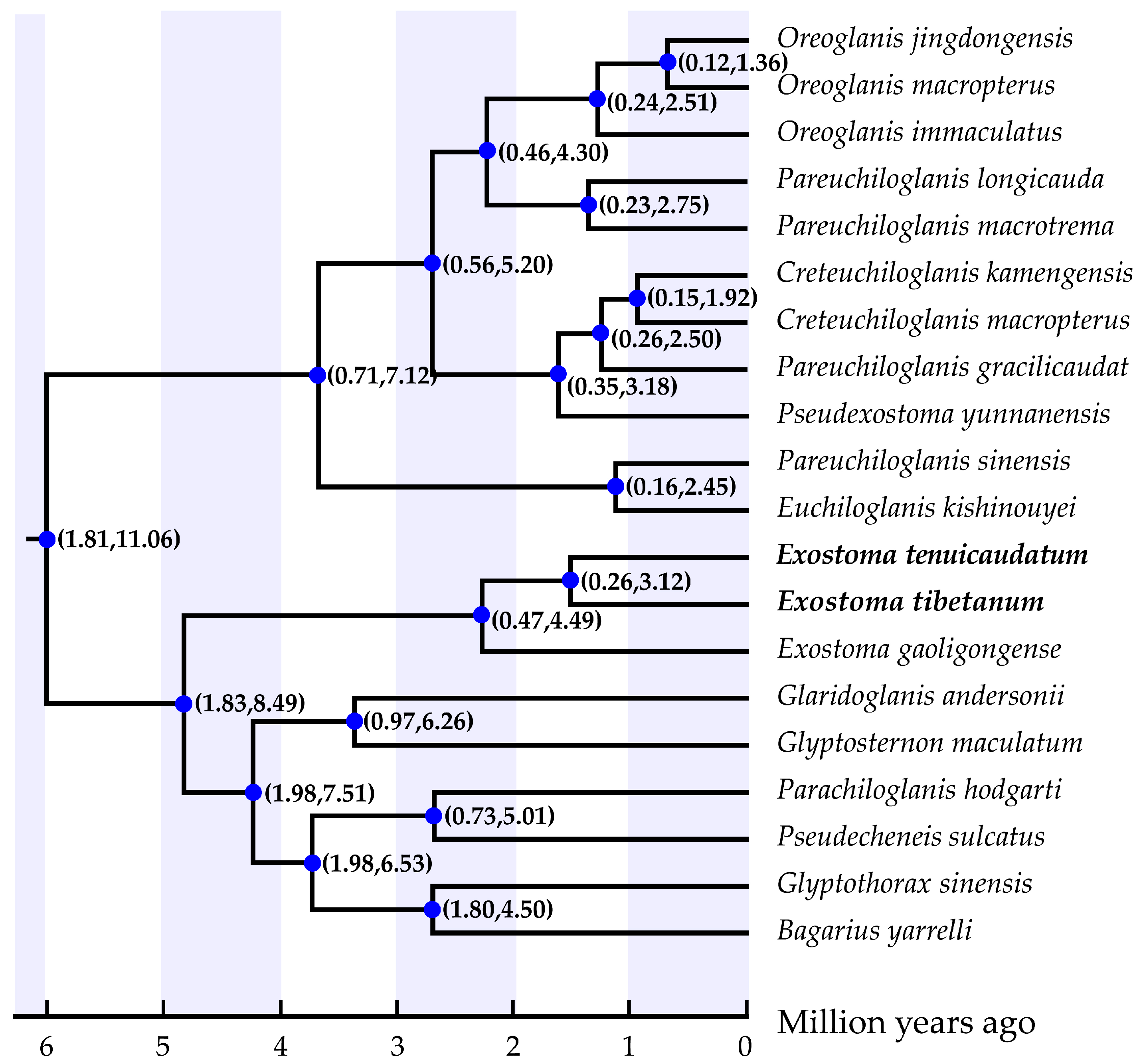
3.3. Phylogenetic Analysis and Divergence Time Estimation
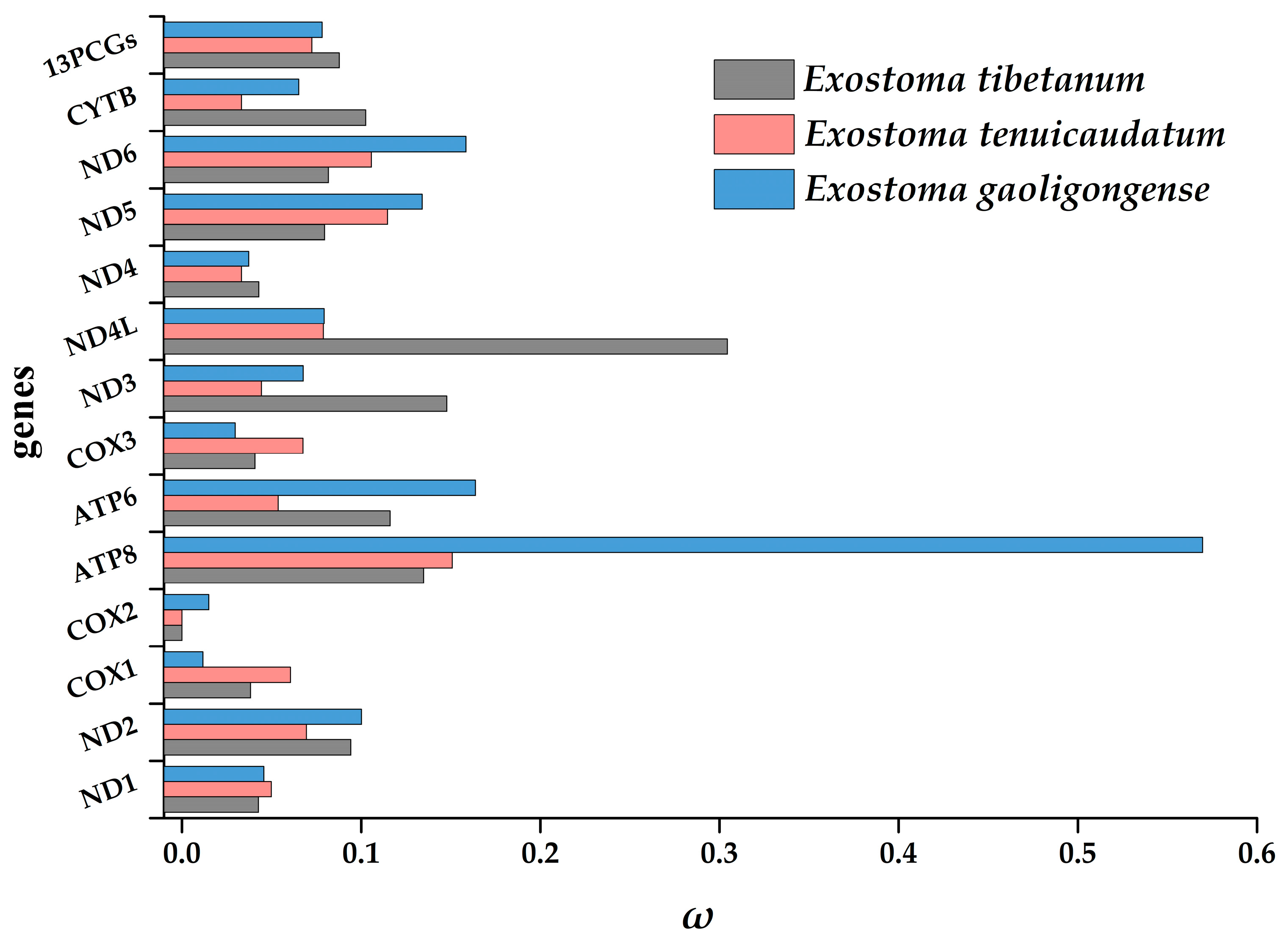
3.4. Selection Pressure Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Satoh, T.P.; Miya, M.; Mabuchi, K.; Nishida, N. Structure and variation of the mitochondrial genome of fishes. BMC Genom. 2016, 17, 719. [Google Scholar] [CrossRef]
- Smith, D.R. The past, present and future of mitochondrial genomics: Have we sequenced enough mtDNAs? Brief. Funct. Genom. 2016, 15, 47–54. [Google Scholar] [CrossRef]
- Vasileiou, P.V.S.; Mourouzis, I.; Pantos, C. Principal aspects regarding the maintenance of mammalian mitochondrial genome integrity. Int. J. Mol. Sci. 2017, 18, 1821. [Google Scholar] [CrossRef]
- Tamang, L.; Sinha, B.; Gurumayum, S.D. Exostoma tenuicaudata, a new species of glyptosternine catfish (Siluriformes: Sisoridae) from the upper Brahmaputra drainage, northeastern India. Zootaxa 2015, 4048, 441–445. [Google Scholar] [CrossRef]
- Darshan, A.; Vishwanath, W.; Abujam, S.; Das, D.N. Exostoma kottelati, a new species of catfish (Teleostei: Sisoridae) from Arunachal Pradesh, India. Zootaxa 2019, 4585, 369–377. [Google Scholar] [CrossRef]
- Ng, H.H. Exostoma ericinum, a new glyptosternine catfish from southwestern China (Teleostei: Siluriformes: Sisoridae). Zootaxa 2018, 4420, 405–414. [Google Scholar] [CrossRef]
- Fricke, R.; Eschmeyer, W.N.; Van der Laan, R. (Eds.) Eschmeyer’s Catalog of Fishes: Genera, Species, References. Available online: http://researcharchive.calacademy.org/research/ichthyology/catalog/fishcatmain.asp (accessed on 6 June 2022).
- Gong, Z.; Lin, P.; Liu, F.; Liu, H. Exostoma tibetana, a new glyptosternine catfish from the lower Yarlung Tsangpo River drainage in southeastern Tibet, China (Siluriformes: Sisoridae). Zootaxa 2018, 4527, 392–402. [Google Scholar] [CrossRef]
- Gong, Z.; Lin, F.; Luo, Z.; Chen, X. The complete mitochondrial genome of Exostoma gaoligongense (Siluriformes: Sisoridae) and its phylogenetic analysis within glyptosternine catfishes. Mitochondrial DNA B 2021, 6, 1424–1425. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Meng, G.; Li, Y.; Yang, C.; Liu, S. MitoZ: A toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 2019, 47, e63. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Stothard, P.; Wishart, D.S. Circular genome visualization and exploration using CGView. Bioinformatics 2005, 21, 537–539. [Google Scholar] [CrossRef]
- Xia, X.H. DAMBE6: New tools for microbial genomics, phylogenetics and molecular evolution. J. HERED. 2017, 108, 431–437. [Google Scholar] [CrossRef]
- Shimodaira, H.; Hasegawa, M. Multiple comparisons of Log Likelihoods with applications to phylogenetic inference. Mol. Biol. Evol. 1999, 16, 1114–1116. [Google Scholar] [CrossRef]
- Nadimi, M.; Daubois, L.; Hijri, M. Mitochondrial comparative genomics and phylogenetic signal assessment of mtDNA among arbuscular mycorrhizal fungi. Mol. Phylogenet. Evol. 2016, 98, 74–83. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlic, I.; Zou, H.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Ronquist, K.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Phylogenetics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Andrew, R. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef]
- Lydekker, R. Indian Tertiary and Post-Tertiary Vertebrata. Tertiary Fishes. Palaeontologia Indica 1886, 10, 241–258. [Google Scholar]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J. TreeAnnotator v1.8.2: MCMC Output Analysis. 2015. Available online: http://beast.bio.ed.ac.uk (accessed on 1 June 2022).
- Rambaut, A. FigTree v1.4.2: Tree Figure Drawing Tool. 2014. Available online: http://tree.bio.ed.ac.uk (accessed on 1 June 2022).
- Yang, Z.; Bielawski, J.P. Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 2000, 15, 496–503. [Google Scholar] [CrossRef]
- Nei, M. Selectionism and neutralism in molecular evolution. Mol. Biol. Evol. 2005, 22, 2318–2342. [Google Scholar] [CrossRef]
- Gao, F.; Chen, C.; Arab, D.A.; Du, Z.; He, Y.; Ho, S.Y.W. EasyCodeML: A visual tool for analysis of selection using CodeML. Ecol. Evol. 2019, 9, 3891–3898. [Google Scholar] [CrossRef]
- Prosdocimi, F.; Carvalho, D.; Beheregaray, A. The complete mitochondrial genome of two recently derived species of the fish genus Nannoperca (Perciformes, Percichthyidae). Mol. Biol. Rep. 2012, 39, 2767–2772. [Google Scholar] [CrossRef]
- Lv, W.; Jiang, H.; Bo, J.; Wang, C.; Yang, L.; He, S. Comparative mitochondrial genome analysis of Neodontobutis hainanensis and Perccottus glenii reveals conserved genome organization and phylogeny. Genomics 2020, 112, 3862–3870. [Google Scholar] [CrossRef]
- Wu, N.; Liu, J.; Wang, S.; Guo, X. Comparative analysis of mitochondrial genomes in two subspecies of the sunwatcher toad-headed agama (Phrynocephalus helioscopus): Prevalent intraspecific gene rearrangements in phrynocephalus. Genes 2022, 13, 203. [Google Scholar] [CrossRef]
- Han, C.; Li, Q.; Xu, J.; Huang, J. Characterization of Clarias gariepinus mitochondrial genome sequence and a comparative analysis with other catfishes. Biologia 2015, 70, 1245–1253. [Google Scholar] [CrossRef]
- Ma, X.; Kang, J.; Chen, W.; Zhou, C.; He, S. Biogeographic history and high-elevation adaptations inferred from the mitochondrial genome of Glyptosternoid fishes (Sisoridae, Siluriformes) from the southeastern Tibetan Plateau. BMC Evol. Biol. 2015, 15, 233. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wu, P.D.; Zhang, D.Z.; Zhang, H.B.; Tang, B.T.; Liu, Q.N.; Dai, L.S. Mitochondrial genome of the yellow catfish Pelteobagrus fulvidraco and insights into Bagridae phylogenetics. Genomics 2019, 111, 1258–1265. [Google Scholar] [CrossRef]
- Chen, W.; Qian, W.; Miao, K.; Qian, R.; Yuan, S.; Liu, W.; Dai, J.; Hu, C.; Chang, Q. Comparative mitogenomics of true frogs (Ranidae, Anura), and its implications for the phylogeny and evolutionary history of Rana. Animals 2022, 12, 1250. [Google Scholar] [CrossRef]
- Yu, P.; Zhou, L.; Yang, W.-T.; Miao, L.-J.; Li, Z.; Zhang, X.-J.; Wang, Y.; Gui, J.-F. Comparative mitogenome analyses uncover mitogenome features and phylogenetic implications of the subfamily Cobitinae. BMC Genomics 2021, 22, 50. [Google Scholar] [CrossRef]
- Chai, H.N.; Du, Y.Z. The complete mitochondrial genome of the pink stem borer, Sesamia inferens, in comparison with four other noctuid moths. Int. J. Mol. Sci. 2012, 13, 10236–10256. [Google Scholar] [CrossRef]
- Wang, C.; Ye, P.; Liu, M.; Zhang, Y.; Feng, H.; Liu, J.; Zhou, H.; Wang, J.; Chen, X. Comparative analysis of four complete mitochondrial genomes of Epinephelidae (Perciformes). Genes 2022, 13, 660. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Cao, C.; Chen, Y. The uplift of Qinghai-Xizang (Tibet) Plateau and the vicariance speciation of glyptosteriod fishes (Siluriformes: Sisoridae). Sci. China Ser. C 2001, 31, 185–192. [Google Scholar]
- Peng, Z.; Ho, S.Y.; Zhang, Y.; He, S. Uplift of the Tibetan plateau: Evidence from divergence times of glyptosternoid catfishes. Mol Phylogenet Evol. 2006, 39, 568–572. [Google Scholar] [CrossRef]
- Clark, M.K.; Schoenbohm, L.M.; Royden, L.H.; Whipple, K.X.; Burchfiel, B.C.; Zhang, X.; Tang, W.; Wang, E.; Chen, L. Surface uplift, tectonics, and erosion of eastern Tibet from large-scale drainage patterns. Tectonics 2004, 23, TC1006. [Google Scholar] [CrossRef]
- Brookfield, M. The evolution of the great river systems of southern Asia during the Cenozoic India-Asia collision: Rivers draining southwards. Geomorphology 1998, 22, 285–312. [Google Scholar] [CrossRef]
- Zeitler, P.K.; Meltzer, A.S.; Koons, P.O.; Craw, D.; Hallet, B.; Chamberlain, C.P.; Kidd, W.S.F.; Park, S.K.; Seeber, L.; Bishop, M. Erosion, Himalayan geodynamics, and the geomorphology of metamorphism. GSA Today 2001, 11, 4–9. [Google Scholar] [CrossRef]
- Meiklejohn, C.D.; Montooth, K.L.; Rand, D.M. Positive and negative selection on the mitochondrial genome. Trends Genet. 2007, 23, 259–263. [Google Scholar] [CrossRef] [PubMed]
- García, G.O.; Oteo, J.A. Evolutionary distances corrected for purifying selection and ancestral polymorphisms. J. Theor. Boil. 2019, 483, 110004. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wang, H.; Yang, L.; Liu, S.; Zhuang, Z. Complete Mitochondrial Genome of Pseudocaranx dentex (Carangidae, Perciformes) Provides Insight into Phylogenetic and Evolutionary Relationship among Carangidae Family. Genes 2021, 12, 1234. [Google Scholar] [CrossRef]
- Li, Q.; Wang, Q.; Chen, C.; Jin, X.; Chen, Z.; Xiong, C.; Li, P.; Zhao, J.; Huang, W. Characterization and comparative mitogenomic analysis of six newly sequenced mitochondrial genomes from ectomycorrhizal fungi (Russula) and phylogenetic analysis of the Agaricomycetes. Int. J. Boil. Macromol. 2018, 119, 792–802. [Google Scholar] [CrossRef]
- Zhang, D.-L.; Gao, J.; Li, M.; Yuan, J.; Liang, J.; Yang, H.; Bu, W. The complete mitochondrial genome of Tetraphleps aterrimus (Hemiptera: Anthocoridae): Genomic comparisons and phylogenetic analysis of Cimicomorpha. Int. J. Boil. Macromol. 2019, 130, 369–377. [Google Scholar] [CrossRef]
- Li, Z.; Li, M.; Xu, S.; Liu, L.; Chen, Z.; Zou, K. Complete Mitogenomes of Three Carangidae (Perciformes) Fishes: Genome Description and Phylogenetic Considerations. Int. J. Mol. Sci. 2020, 21, 4685. [Google Scholar] [CrossRef]
- Dan, B.Y.; Blumberg, A.; Dan, M. Mitochondrial-nuclear co-evolution and its effects on OXPHOS activity and regulation. Biochim. Biophys. Acta. 2012, 1819, 1107–1111. [Google Scholar]
- Garvin, M.R.; Bielawski, J.P.; Sazanov, L.A.; Gharrett, A.J. Review and meta-analysis of natural selection in mitochondrial complex I in metazoans. J. Zool. Syst. Evol. Res. 2015, 53, 1–17. [Google Scholar] [CrossRef]
- Caballero, S.; Duchene, S.; Garavito, M.F.; Slikas, B.; Baker, C.S. Initial evidence for adaptive selection on the NADH subunit two of freshwater dolphins by analyses of mitochondrial genomes. PLoS ONE 2015, 10, e0123543. [Google Scholar] [CrossRef]
- Consuegra, S.; John, E.; Verspoor, E.; De Leaniz, C.G. Patterns of natural selection acting on the mitochondrial genome of a locally adapted fish species. Genet. Sel. Evol. 2015, 47, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wang, X.; Ting, N.; Zhang, Y. Mitogenomic analysis of Chinese snub-nosed monkeys: Evidence of positive selection in NADH dehydrogenase genes in high-altitude adaptation. Mitochondrion 2011, 11, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Chen, Y.; Liu, F.; Gao, Y. Mitochondrial genome of Tibetan wild ass (Equus kiang) reveals substitutions in NADH which may reflect evolutionary adaptation to cold and hypoxic conditions. Asia Life Sci. 2012, 21, 1–11. [Google Scholar]
- Wang, Y.; Shen, Y.; Feng, C.; Zhao, K.; Song, Z.; Zhang, Y.; Yang, L.; He, S. Mitogenomic perspectives on the origin of Tibetan loaches and their adaptation to high altitude. Sci. Rep. 2016, 6, 29690. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Drainage | GenBank No. | Size (bp) |
|---|---|---|---|
| Bagarius yarrelli | Irrawaddy River | NC021606 | 16,503 |
| Creteuchiloglanis kamengensis | Yarlung Tsangpo River | MN396886 | 16,589 |
| Creteuchiloglanis macropterus | Nujiang River | KP872683 | 16,589 |
| Euchiloglanis kishinouyei | Jinsha River | NC021598 | 16,561 |
| Exostoma gaoligongense | Nujiang River | NC056351 | 16,529 |
| Exostoma tibetanum | Yarlung Tsangpo River | ON641840 | 16,528 |
| Exostoma tenuicaudatum | Yarlung Tsangpo River | ON641841 | 16,533 |
| Glaridoglanis andersonii | Lohit River | NC021600 | 16,532 |
| Glyptosternon maculatum | Yarlung Tsangpo River | NC021597 | 16,539 |
| Glyptothorax sinensis | Yangtze River | KJ739617 | 16,531 |
| Oreoglanis immaculatus | Irrawaddy River | NC028511 | 16,576 |
| Oreoglanis jingdongensis | Irrawaddy River | NC028512 | 16,569 |
| Oreoglanis macropterus | Nujiang River | NC021607 | 16,568 |
| Parachiloglanis hodgarti | Yarlung Tsangpo River | MW715684 | 16,511 |
| Pareuchiloglanis gracilicaudata | Lancang River | NC021603 | 16,588 |
| Pareuchiloglanis longicauda | Pearl River | NC028514 | 16,535 |
| Pareuchiloglanis macrotrema | Red River | NC028515 | 16,570 |
| Pareuchiloglanis sinensis | Jinsha River | KP872695 | 16,572 |
| Pseudecheneis sulcatus | Yarlung Tsangpo River | MK843301 | 16,535 |
| Pseudexostoma yunnanensis | Irrawaddy River | JQ026258 | 16,598 |
| A + T (%) | G + C (%) | AT Skewness | GC Skewness | |||||
|---|---|---|---|---|---|---|---|---|
| ti | te | ti | te | ti | te | ti | te | |
| Whole mitogenome | 54.8 | 55.2 | 45.2 | 44.8 | 0.135 | 0.138 | –0.256 | –0.315 |
| Protein-coding genes | 54.4 | 54.8 | 45.6 | 45.2 | 0.058 | 0.066 | –0.325 | –0.330 |
| 1st codon position | 48.1 | 48.1 | 51.9 | 51.9 | 0.152 | 0.168 | –0.042 | –0.050 |
| 2nd codon position | 58.5 | 58.7 | 41.5 | 41.3 | –0.371 | –0.365 | –0.354 | –0.363 |
| 3rd codon position | 56.6 | 57.7 | 43.4 | 42.3 | 0.432 | 0.411 | –0.631 | –0.641 |
| rRNA genes | 53.8 | 54.4 | 46.2 | 45.6 | 0.279 | 0.283 | –0.156 | –0.155 |
| tRNA genes | 55.6 | 56.4 | 44.4 | 43.6 | 0.065 | 0.046 | 0.009 | –0.002 |
| Control region | 62.4 | 59.6 | 37.6 | 40.4 | 0.071 | 0.013 | –0.186 | –0.094 |
| Genes | Model | −LnL | Df. | Parameter Estimates | p-Value | Positive Sites |
|---|---|---|---|---|---|---|
| ND1 | Fore_ti | −6879.48 | 1 | f0 = 0.966, f1 = 0.030, f2a = 0.004, f2b = 0.000, ω0 = 0.040, ω1 = 1, ω2 = 13.576 | 0.062 | 103 N 0.982 * |
| Null | −6881.22 | 1 | ω0 = 0.040, ω1 = 1, ω2 = 1 | |||
| ND6 | Fore_ga | −3759.45 | 1 | f0 = 0.874, f1 = 0.110, f2a = 0.015, f2b = 0.002, ω0 = 0.044, ω1 = 1, ω2 = 2.740 | 0.030 | 46 H 0.961 * |
| null | −3757.10 | 1 | ω0 = 0.044, ω1 = 1, ω2 = 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, Z.; Jiang, W.; Feng, H.; Liu, Y.; Zhu, T. Comparative Mitogenomics of Two Sympatric Catfishes of Exostoma (Siluriformes: Sisoridae) from the Lower Yarlung Tsangpo River and Its Application for Phylogenetic Consideration. Genes 2022, 13, 1615. https://doi.org/10.3390/genes13091615
Gong Z, Jiang W, Feng H, Liu Y, Zhu T. Comparative Mitogenomics of Two Sympatric Catfishes of Exostoma (Siluriformes: Sisoridae) from the Lower Yarlung Tsangpo River and Its Application for Phylogenetic Consideration. Genes. 2022; 13(9):1615. https://doi.org/10.3390/genes13091615
Chicago/Turabian StyleGong, Zheng, Wanxiang Jiang, Huizhe Feng, Yanchao Liu, and Tianshun Zhu. 2022. "Comparative Mitogenomics of Two Sympatric Catfishes of Exostoma (Siluriformes: Sisoridae) from the Lower Yarlung Tsangpo River and Its Application for Phylogenetic Consideration" Genes 13, no. 9: 1615. https://doi.org/10.3390/genes13091615
APA StyleGong, Z., Jiang, W., Feng, H., Liu, Y., & Zhu, T. (2022). Comparative Mitogenomics of Two Sympatric Catfishes of Exostoma (Siluriformes: Sisoridae) from the Lower Yarlung Tsangpo River and Its Application for Phylogenetic Consideration. Genes, 13(9), 1615. https://doi.org/10.3390/genes13091615