




Genes 2025, 16(4), 390; https://doi.org/10.3390/genes16040390 - 28 Mar 2025
Viewed by 283
Abstract
Background: The presence of light-pigmented facial stripes, parallel on both sides of the cranial region, is a widespread characteristic in various goat breeds of European origin and beyond. In Italy, this phenotype is relatively common from the north to the south of the
[...] Read more.
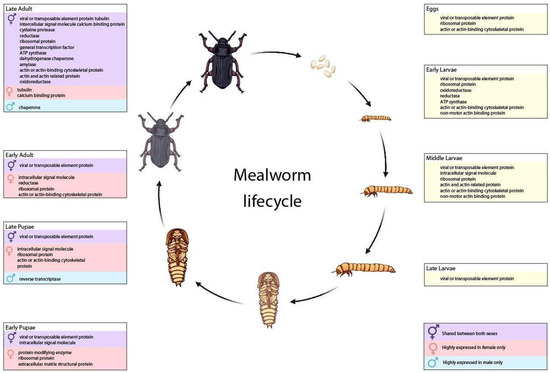
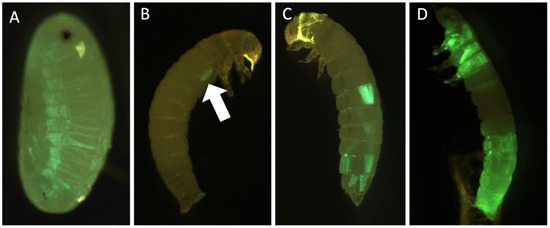
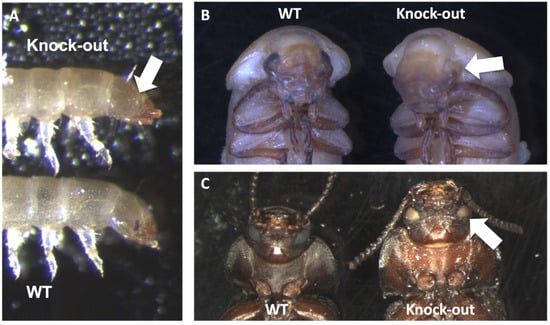
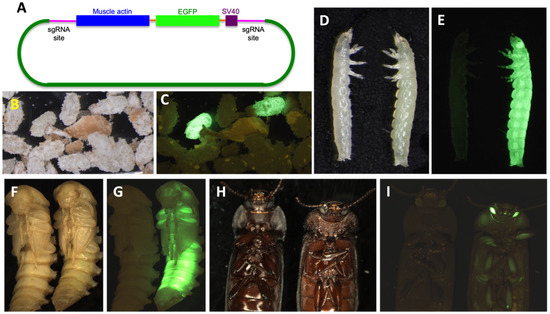
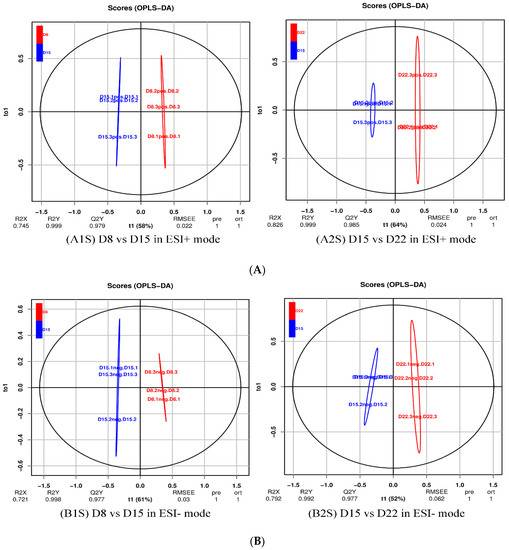
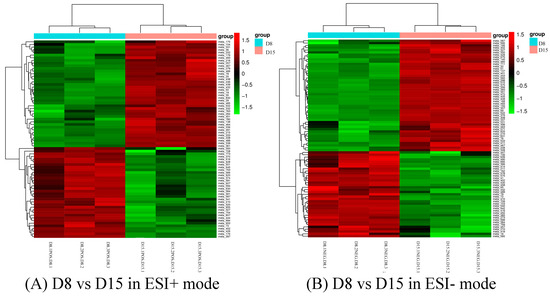
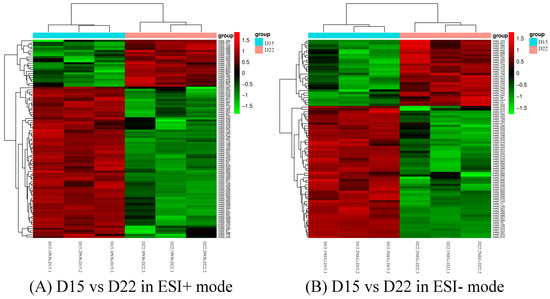
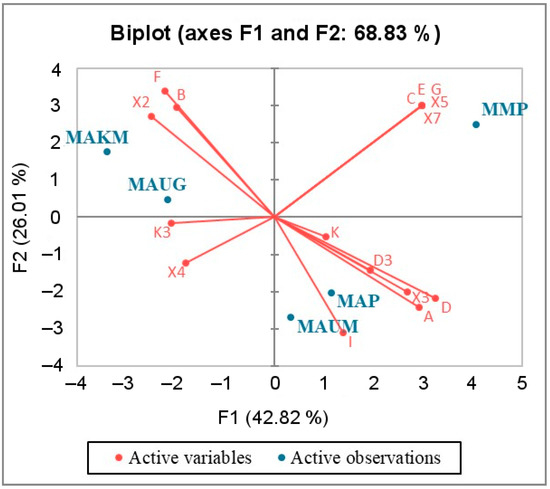
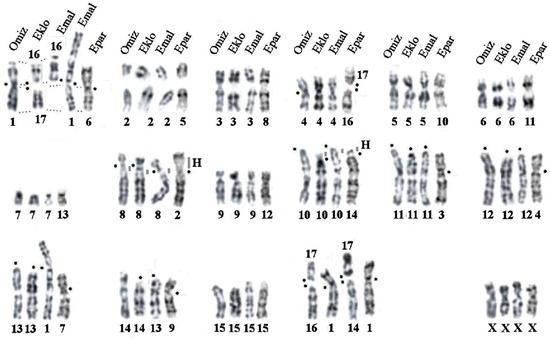
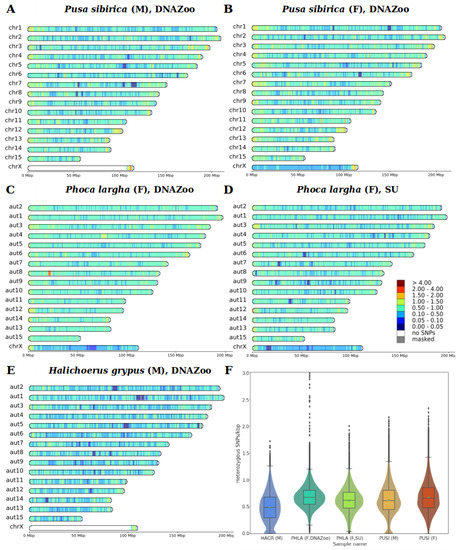
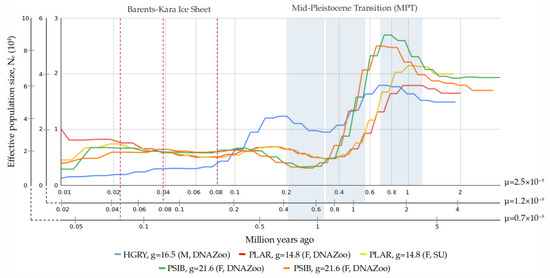
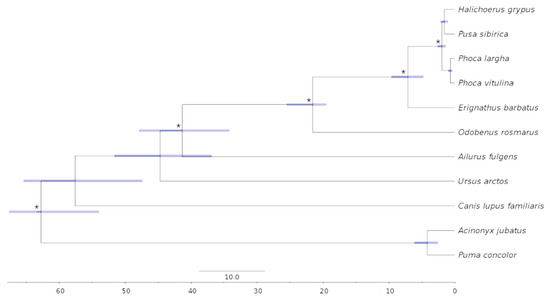
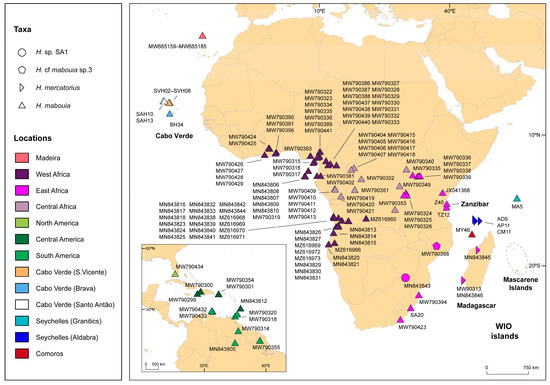
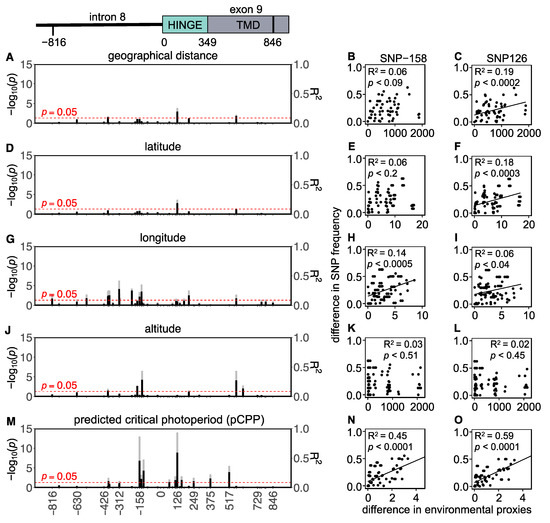
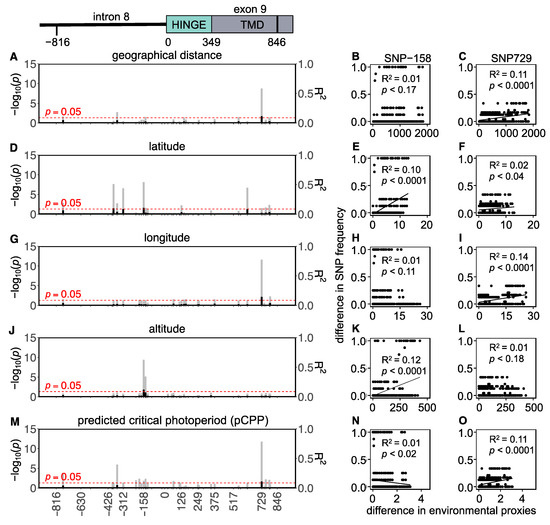
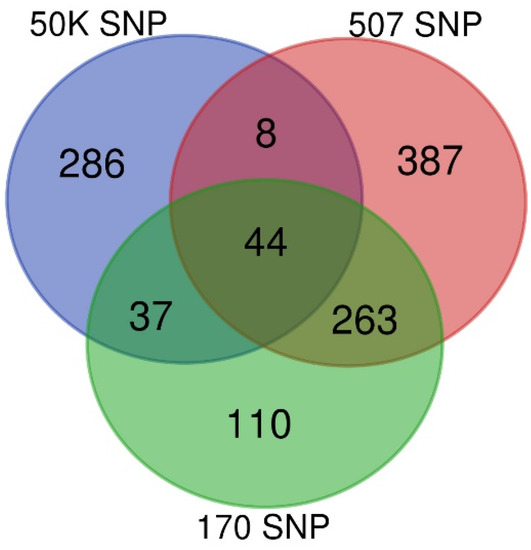
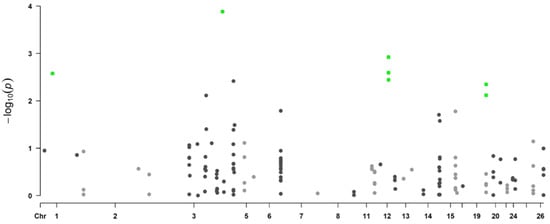
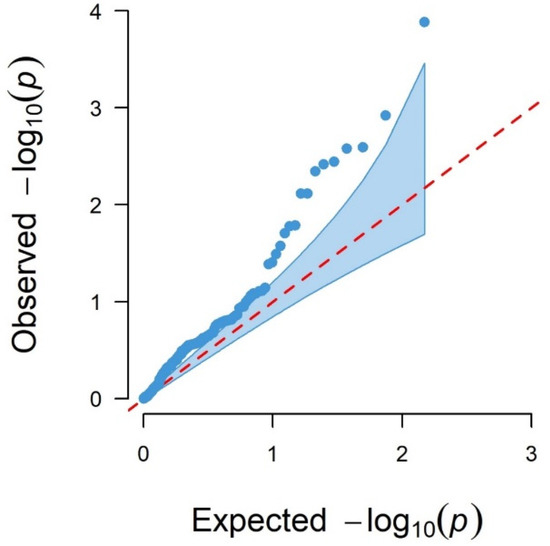
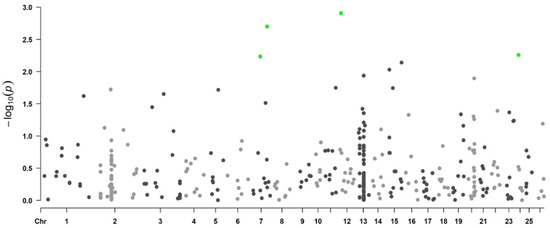
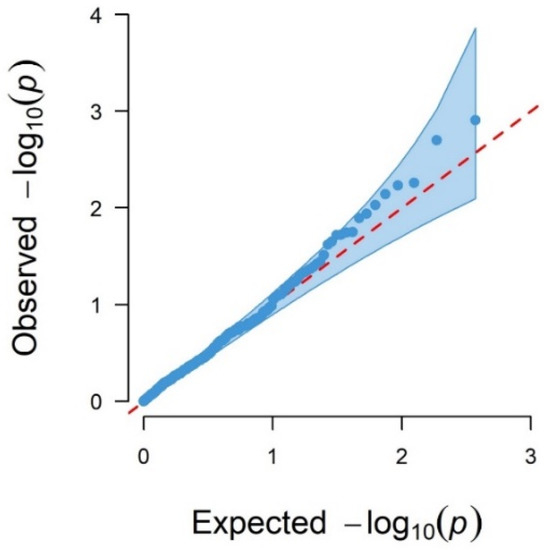
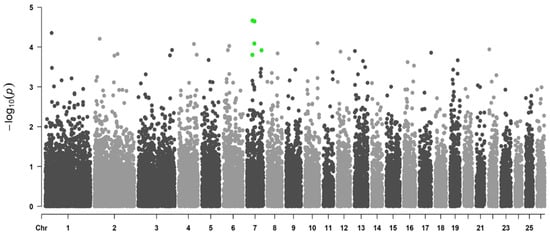
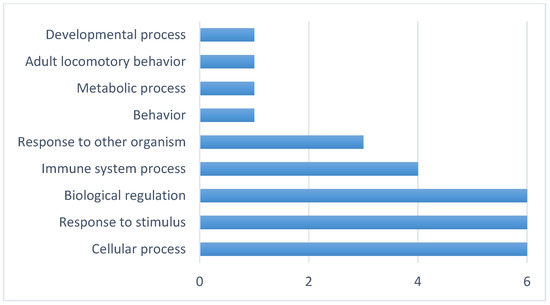
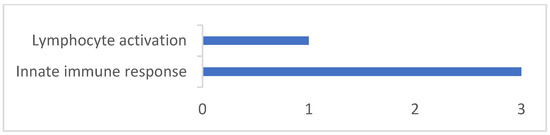
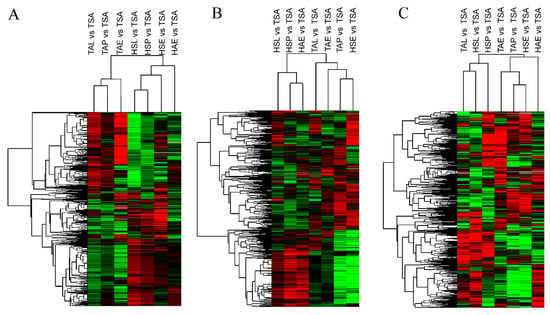
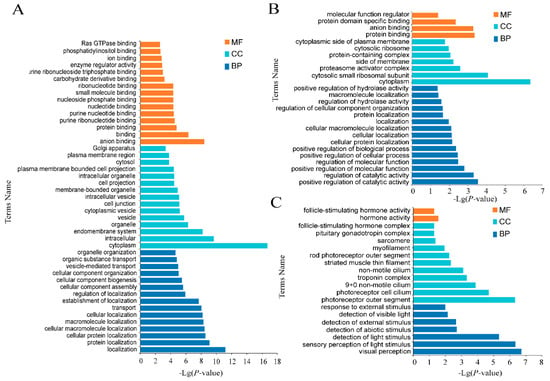
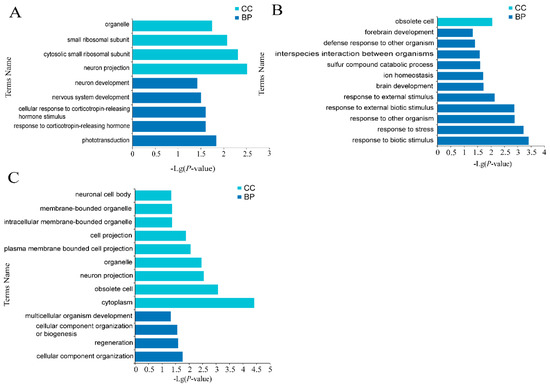
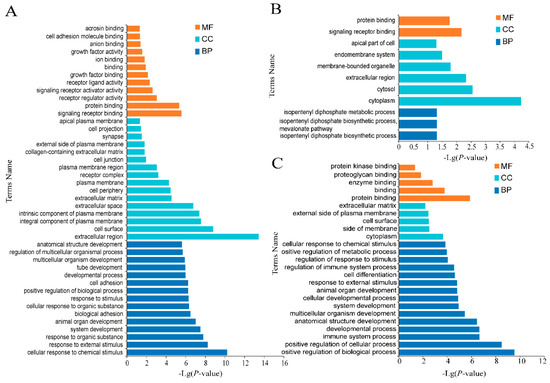
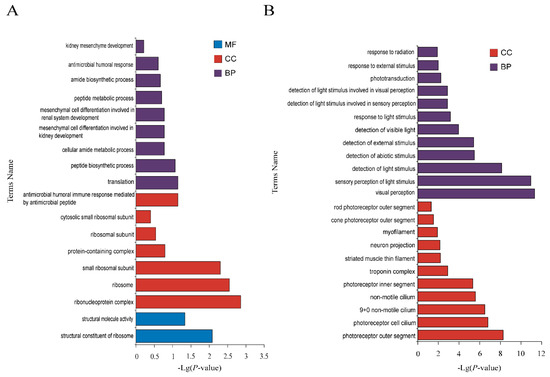
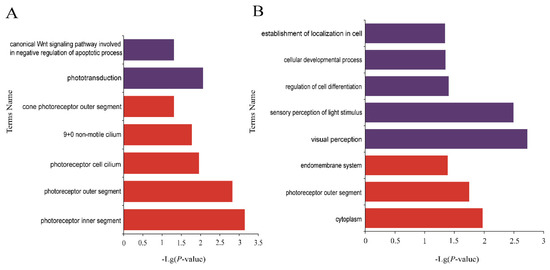
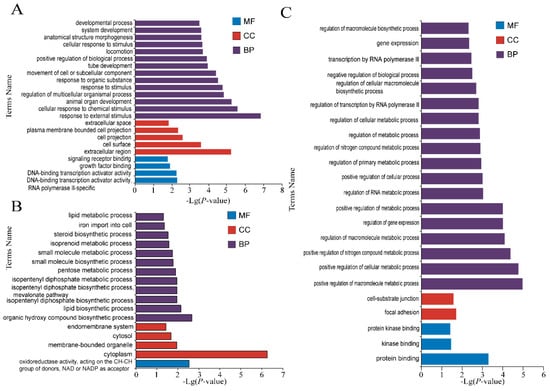
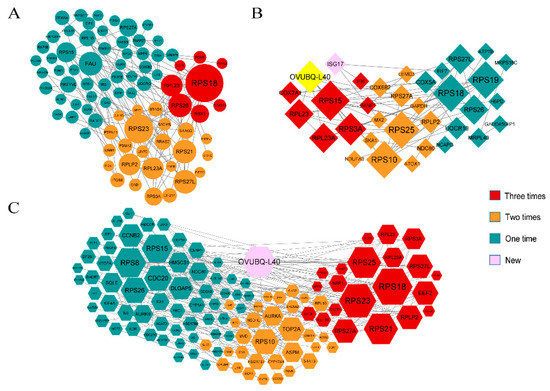
Background: The presence of light-pigmented facial stripes, parallel on both sides of the cranial region, is a widespread characteristic in various goat breeds of European origin and beyond. In Italy, this phenotype is relatively common from the north to the south of the peninsula. The availability of genotypic data at single-nucleotide polymorphism (SNP) loci for breeds and populations characterized by such a pigmentation pattern enabled us to study the genomic regions potentially correlated with this phenotype, for simplicity referred to as “facciuto”. Methods: We adopted an FST-outlier approach to detect signals of differential selection in 18 pairwise comparisons, each involving 6 genetic goat types with the “facciuto” phenotype (Facciuta Lucana, Facciuta della Valnerina, Valfortorina, Teramana, Capestrina, and Roccaverano) contrasted with each of 3 “non-facciuto” goat breeds selected as reference populations (Red Mediterranean, light brown; Saanen, white; Malagueña, mahogany solid). Results: The analysis of the region ±200 kbps upstream and downstream of the two significant signals on chromosome 13 and 15 allowed us to identify, among the annotated genes, ASIP, AHCY, ITCH, DYNLRB1, MAP1LC3A, PIGU, LOC102177263, and DTX4, whose functions could be related to several mechanisms underlying the phenotype under investigation. Conclusions: This study confirmed the fundamental role of ASIP in pigmentation, although additional pathways may concurrently contribute to the determinism of the considered “facciuto” phenotype in Italian goats.
Full article







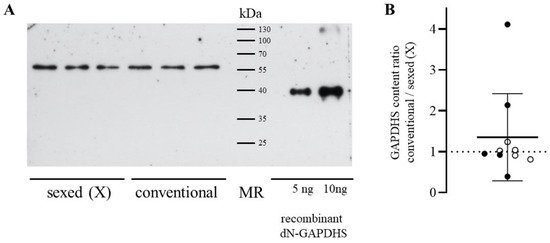
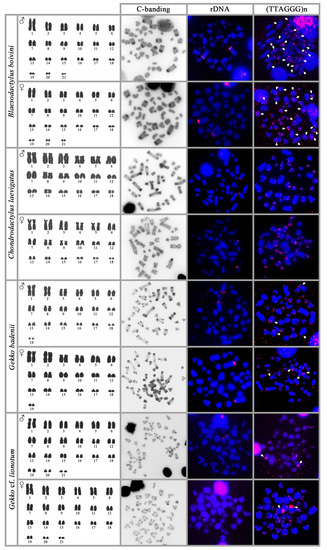
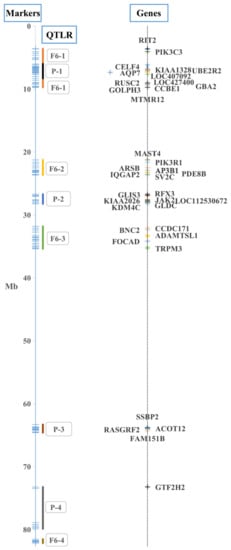
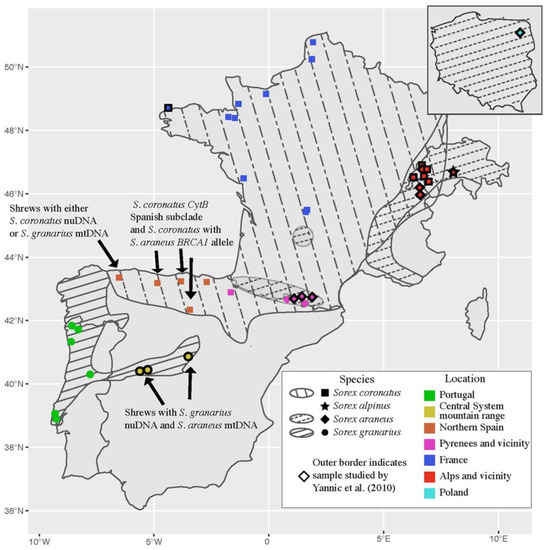




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}