
Genetic Insights from Consanguineous Cardiomyopathy Families

 , , ,
, , ,  ,
,  add
Show full author list
add
Show full author list
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Clinical Characteristics of the Patients
3.2. Genetic Findings
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maron, B.J. Clinical Course and Management of Hypertrophic Cardiomyopathy. N. Engl. J. Med. 2018, 379, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Makavos, G.; Κairis, C.; Tselegkidi, M.E.; Karamitsos, T.; Rigopoulos, A.G.; Noutsias, M.; Ikonomidis, I. Hypertrophic Cardiomyopathy: An Updated Review on Diagnosis, Prognosis, and Treatment. Heart Fail. Rev. 2019, 24, 439–459. [Google Scholar] [CrossRef]
- Maron, B.A.; Wang, R.S.; Carnethon, M.R.; Rowin, E.J.; Loscalzo, J.; Maron, B.J.; Maron, M.S. What Causes Hypertrophic Cardiomyopathy? Am. J. Cardiol. 2022, 179, 74–82. [Google Scholar] [CrossRef]
- Orphanou, N.; Papatheodorou, E.; Anastasakis, A. Dilated Cardiomyopathy in the Era of Precision Medicine: Latest Concepts and Developments. Heart Fail. Rev. 2022, 27, 1173–1191. [Google Scholar] [CrossRef]
- Weintraub, R.G.; Semsarian, C.; Macdonald, P. Dilated Cardiomyopathy. Lancet 2017, 390, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Jordan, E.; Peterson, L.; Ai, T.; Asatryan, B.; Bronicki, L.; Brown, E.; Celeghin, R.; Edwards, M.; Fan, J.; Ingles, J.; et al. Evidence-Based Assessment of Genes in Dilated Cardiomyopathy. Circulation 2021, 144, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Hamamy, H.; Antonarakis, S.E.; Cavalli-Sforza, L.L.; Temtamy, S.; Romeo, G.; Kate, L.P.T.; Bennett, R.L.; Shaw, A.; Megarbane, A.; van Duijn, C.; et al. Consanguineous Marriages, Pearls and Perils: Geneva International Consanguinity Workshop Report. Genet. Med. 2011, 13, 841–847. [Google Scholar] [CrossRef]
- Iuso, A.; Wiersma, M.; Schüller, H.J.; Pode-Shakked, B.; Marek-Yagel, D.; Grigat, M.; Schwarzmayr, T.; Berutti, R.; Alhaddad, B.; Kanon, B.; et al. Mutations in PPCS, Encoding Phosphopantothenoylcysteine Synthetase, Cause Autosomal-Recessive Dilated Cardiomyopathy. Am. J. Hum. Genet. 2018, 102, 1018–1030. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.G.; Mazaheri, N.; Maroofian, R.; Zamani, M.; Seifi, T.; Sedaghat, A.; Shariati, G.; Jamshidi, Y.; Allen, H.D.; Wehrens, X.H.T.; et al. Analysis of Enriched Rare Variants in JPH2-Encoded Junctophilin-2 among Greater Middle Eastern Individuals Reveals a Novel Homozygous Variant Associated with Neonatal Dilated Cardiomyopathy. Sci. Rep. 2019, 9, 9038. [Google Scholar] [CrossRef]
- Osborn, D.P.S.; Emrahi, L.; Clayton, J.; Tabrizi, M.T.; Wan, A.Y.B.; Maroofian, R.; Yazdchi, M.; Garcia, M.L.E.; Galehdari, H.; Hesse, C.; et al. Autosomal Recessive Cardiomyopathy and Sudden Cardiac Death Associated with Variants in MYL3. Genet. Med. 2021, 23, 787–792. [Google Scholar] [CrossRef]
- Hedberg-Oldfors, C.; Abramsson, A.; Osborn, D.P.S.; Danielsson, O.; Fazlinezhad, A.; Nilipour, Y.; Hübbert, L.; Nennesmo, I.; Visuttijai, K.; Bharj, J.; et al. Cardiomyopathy with Lethal Arrhythmias Associated with Inactivation of KLHL24. Hum. Mol. Genet. 2019, 28, 1919–1929. [Google Scholar] [CrossRef]
- Vasilescu, C.; Ojala, T.H.; Brilhante, V.; Ojanen, S.; Hinterding, H.M.; Palin, E.; Alastalo, T.P.; Koskenvuo, J.; Hiippala, A.; Jokinen, E.; et al. Genetic Basis of Severe Childhood-Onset Cardiomyopathies. J. Am. Coll. Cardiol. 2018, 72, 2324–2338. [Google Scholar] [CrossRef] [PubMed]
- Johansson, J.; Frykholm, C.; Ericson, K.; Kazamia, K.; Lindberg, A.; Mulaiese, N.; Falck, G.; Gustafsson, P.E.; Lidéus, S.; Gudmundsson, S.; et al. Loss of Nexilin Function Leads to a Recessive Lethal Fetal Cardiomyopathy Characterized by Cardiomegaly and Endocardial Fibroelastosis. Am. J. Med. Genet. A 2022, 188, 1676–1687. [Google Scholar] [CrossRef]
- Koskenvuo, J.W.; Saarinen, I.; Ahonen, S.; Tommiska, J.; Weckström, S.; Seppälä, E.H.; Tuupanen, S.; Kangas-Kontio, T.; Schleit, J.; Heliö, K.; et al. Biallelic Loss-of-Function in NRAP Is a Cause of Recessive Dilated Cardiomyopathy. PLoS ONE 2021, 16, e0245681. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Al-Hassnan, Z.N.; Almesned, A.; Tulbah, S.; Alakhfash, A.; Alhadeq, F.; Alruwaili, N.; Alkorashy, M.; Alhashem, A.; Alrashdan, A.; Faqeih, E.; et al. Categorized Genetic Analysis in Childhood-Onset Cardiomyopathy. Circ. Genom. Precis. Med. 2020, 13, 504–514. [Google Scholar] [CrossRef]
- Monies, D.; Abouelhoda, M.; AlSayed, M.; Alhassnan, Z.; Alotaibi, M.; Kayyali, H.; Al-Owain, M.; Shah, A.; Rahbeeni, Z.; Al-Muhaizea, M.A.; et al. The Landscape of Genetic Diseases in Saudi Arabia Based on the First 1000 Diagnostic Panels and Exomes. Hum. Genet. 2017, 136, 921–939. [Google Scholar] [CrossRef]
- Alfares, A.A.; Kelly, M.A.; McDermott, G.; Funke, B.H.; Lebo, M.S.; Baxter, S.B.; Shen, J.; McLaughlin, H.M.; Clark, E.H.; Babb, L.J.; et al. Results of Clinical Genetic Testing of 2912 Probands with Hypertrophic Cardiomyopathy: Expanded Panels Offer Limited Additional Sensitivity. Genet. Med. 2015, 17, 880–888. [Google Scholar] [CrossRef]
- Hathaway, J.; Heliö, K.; Saarinen, I.; Tallila, J.; Seppälä, E.H.; Tuupanen, S.; Turpeinen, H.; Kangas-Kontio, T.; Schleit, J.; Tommiska, J.; et al. Diagnostic Yield of Genetic Testing in a Heterogeneous Cohort of 1376 HCM Patients. BMC Cardiovasc. Disord. 2021, 21, 126. [Google Scholar] [CrossRef] [PubMed]
- Alazami, A.M.; Patel, N.; Shamseldin, H.E.; Anazi, S.; Al-Dosari, M.S.; Alzahrani, F.; Hijazi, H.; Alshammari, M.; Aldahmesh, M.A.; Salih, M.A.; et al. Accelerating Novel Candidate Gene Discovery in Neurogenetic Disorders via Whole-Exome Sequencing of Prescreened Multiplex Consanguineous Families. Cell Rep. 2015, 10, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Hengel, H.; Buchert, R.; Sturm, M.; Haack, T.B.; Schelling, Y.; Mahajnah, M.; Sharkia, R.; Azem, A.; Balousha, G.; Ghanem, Z.; et al. First-Line Exome Sequencing in Palestinian and Israeli Arabs with Neurological Disorders Is Efficient and Facilitates Disease Gene Discovery. Eur. J. Hum. Genet. 2020, 28, 1034–1043. [Google Scholar] [CrossRef]
- Wagner, M.; Osborn, D.P.S.; Gehweiler, I.; Nagel, M.; Ulmer, U.; Bakhtiari, S.; Amouri, R.; Boostani, R.; Hentati, F.; Hockley, M.M.; et al. Bi-Allelic Variants in RNF170 Are Associated with Hereditary Spastic Paraplegia. Nat. Commun. 2019, 10, 4790. [Google Scholar] [CrossRef] [PubMed]
- Osborn, D.P.S.; Pond, H.L.; Mazaheri, N.; Dejardin, J.; Munn, C.J.; Mushref, K.; Cauley, E.S.; Moroni, I.; Pasanisi, M.B.; Sellars, E.A.; et al. Mutations in INPP5K Cause a Form of Congenital Muscular Dystrophy Overlapping Marinesco-Sjögren Syndrome and Dystroglycanopathy. Am. J. Hum. Genet. 2017, 100, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Truszkowska, G.T.; Bilińska, Z.T.; Muchowicz, A.; Pollak, A.; Biernacka, A.; Kozar-Kamińska, K.; Stawiński, P.; Gasperowicz, P.; Kosińska, J.; Zieliński, T.; et al. Homozygous Truncating Mutation in NRAP Gene Identified by Whole Exome Sequencing in a Patient with Dilated Cardiomyopathy. Sci. Rep. 2017, 7, 3362. [Google Scholar] [CrossRef]
- Mosavi, L.K.; Cammett, T.J.; Desrosiers, D.C.; Peng, Z. The Ankyrin Repeat as Molecular Architecture for Protein Recognition. Protein Sci. 2004, 13, 1435–1448. [Google Scholar] [CrossRef]
- Ramzan, S.; Tennstedt, S.; Tariq, M.; Khan, S.; Noor Ul Ayan, H.; Ali, A.; Munz, M.; Thiele, H.; Korejo, A.A.; Mughal, A.R.; et al. A Novel Missense Mutation in Tnni3k Causes Recessively Inherited Cardiac Conduction Disease in a Consanguineous Pakistani Family. Genes 2021, 12, 1282. [Google Scholar] [CrossRef]
- He, Y.; Maier, K.; Leppert, J.; Hausser, I.; Schwieger-Briel, A.; Weibel, L.; Theiler, M.; Kiritsi, D.; Busch, H.; Boerries, M.; et al. Monoallelic Mutations in the Translation Initiation Codon of KLHL24 Cause Skin Fragility. Am. J. Hum. Genet. 2016, 99, 1395–1404. [Google Scholar] [CrossRef]
- Nilsson, J.; Schoser, B.; Laforet, P.; Kalev, O.; Lindberg, C.; Romero, N.B.; Dávila Lõpez, M.; Akman, H.O.; Wahbi, K.; Iglseder, S.; et al. Polyglucosan Body Myopathy Caused by Defective Ubiquitin Ligase RBCK1. Ann. Neurol. 2013, 74, 914–919. [Google Scholar] [CrossRef]
- Al-Owain, M.; Wakil, S.; Shareef, F.; Al-Fatani, A.; Hamadah, E.; Haider, M.; Al-Hindi, H.; Awaji, A.; Khalifa, O.; Baz, B.; et al. Novel Homozygous Mutation in DSP Causing Skin Fragility-Woolly Hair Syndrome: Report of a Large Family and Review of the Desmoplakin-Related Phenotypes. Clin. Genet. 2011, 80, 50–58. [Google Scholar] [CrossRef]
- Alcalai, R.; Metzger, S.; Rosenheck, S.; Meiner, V.; Chajek-Shaul, T. A Recessive Mutation in Desmoplakin Causes Arrhythmogenic Right Ventricular Dysplasia, Skin Disorder, and Woolly Hair. J. Am. Coll. Cardiol. 2003, 42, 319–327. [Google Scholar] [CrossRef]
- Molho-Pessach, V.; Sheffer, S.; Siam, R.; Tams, S.; Siam, I.; Awwad, R.; Babay, S.; Golender, J.; Simanovsky, N.; Ramot, Y.; et al. Two Novel Homozygous Desmoplakin Mutations in Carvajal Syndrome. Pediatr. Derm. 2015, 32, 641–646. [Google Scholar] [CrossRef]
- Al-Sabeq, B.; Krahn, A.D.; Conacher, S.; Klein, G.J.; Laksman, Z. Arrhythmogenic Right Ventricular Cardiomyopathy with Recessive Inheritance Related to a New Homozygous Desmocollin-2 Mutation. Can. J. Cardiol. 2014, 30, 696.e1–696.e3. [Google Scholar] [CrossRef]
- Simpson, M.A.; Mansour, S.; Ahnood, D.; Kalidas, K.; Patton, M.A.; McKenna, W.J.; Behr, E.R.; Crosby, A.H. Homozygous Mutation of Desmocollin-2 in Arrhythmogenic Right Ventricular Cardiomyopathy with Mild Palmoplantar Keratoderma and Woolly Hair. Cardiology 2009, 113, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Klauke, B.; Kossmann, S.; Gaertner, A.; Brand, K.; Stork, I.; Brodehl, A.; Dieding, M.; Walhorn, V.; Anselmetti, D.; Gerdes, D.; et al. De Novo Desmin-Mutation N116S Is Associated with Arrhythmogenic Right Ventricular Cardiomyopathy. Hum. Mol. Genet. 2010, 19, 4595–4607. [Google Scholar] [CrossRef] [PubMed]
- Gerull, B.; Kirchner, F.; Chong, J.X.; Tagoe, J.; Chandrasekharan, K.; Strohm, O.; Waggoner, D.; Ober, C.; Duff, H.J. Homozygous Founder Mutation in Desmocollin-2 (DSC2) Causes Arrhythmogenic Cardiomyopathy in the Hutterite Population. Circ. Cardiovasc. Genet. 2013, 6, 327–336. [Google Scholar] [CrossRef]
- Lorenzon, A.; Pilichou, K.; Rigato, I.; Vazza, G.; de Bortoli, M.; Calore, M.; Occhi, G.; Carturan, E.; Lazzarini, E.; Cason, M.; et al. Homozygous Desmocollin-2 Mutations and Arrhythmogenic Cardiomyopathy. Am. J. Cardiol. 2015, 116, 1245–1251. [Google Scholar] [CrossRef] [PubMed]


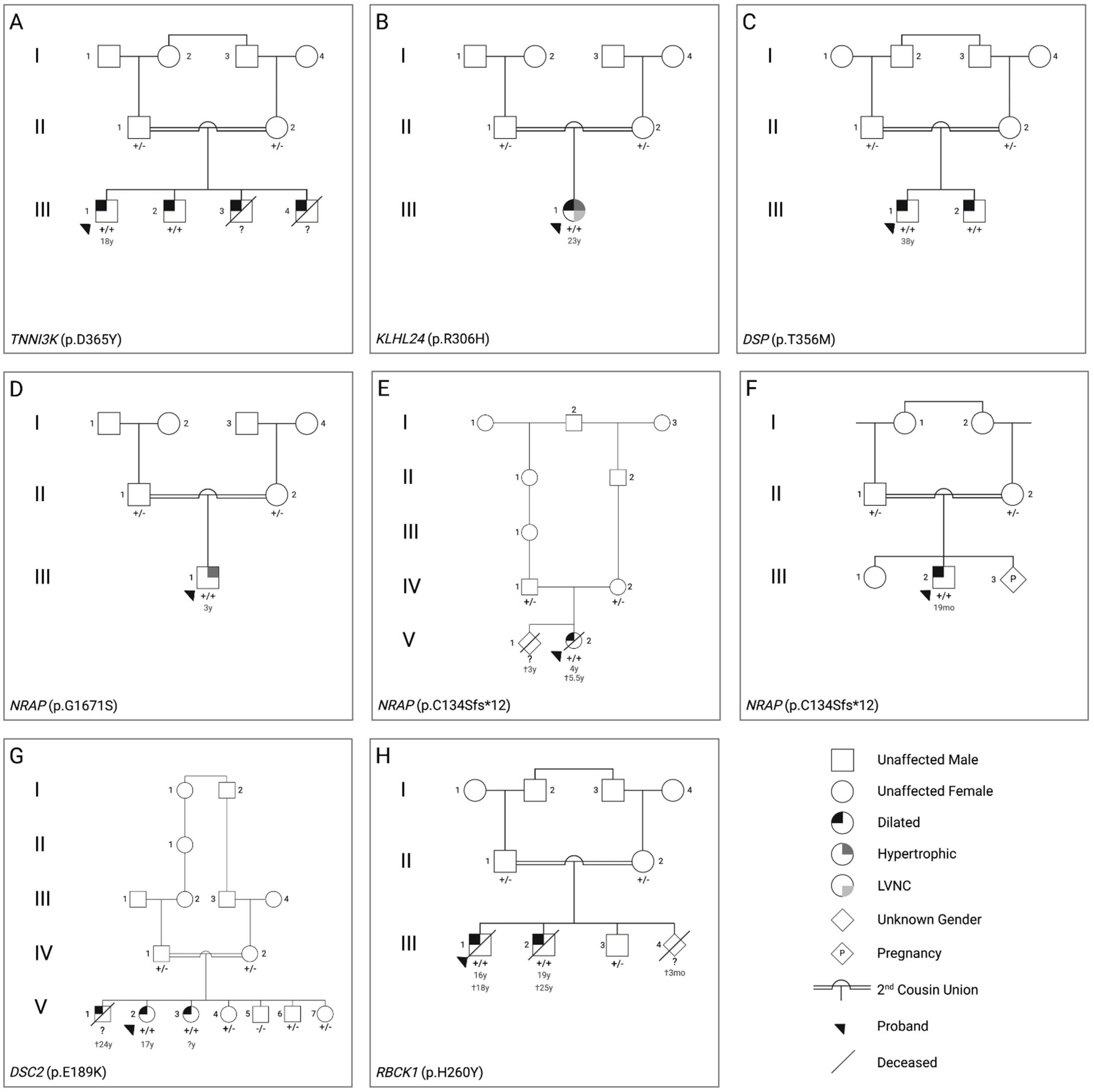{kind=link}
| Family | Sex | Genotype | Phenotype | Proband Imaging | Proband ECG | Other |
|---|---|---|---|---|---|---|
| A | Male | TNNI3K (p.D365Y) | DCM | Mild diastolic dysfunction progressing to moderate/severe with global hypokinesia, LA enlargement, mild billowing of MV with trivial MR, mild TR, mild PR | Left axis deviation, criteria for LA enlargement, intraventricular conduction delay, pre-excitation | Holter: NSVT |
| B | Female | KLHL24 (p.R306H) | Mixed: HCM/DCM with LVNC | Biventricular hypertrophy (LVMWT 28 mm), severe LV enlargement, severe LV systolic dysfunction, moderate diastolic dysfunction, mild LA enlargement | Right axis deviation, RBBB | |
| C | Male | DSP (p.T356M) | DCM | Mild RV enlargement, systolic dysfunction of RV, mild diastolic dysfunction, mild MR and TR | Intraventricular conduction delay, repolarization abnormalities | |
| D | Male | NRAP (p.G1671S) | HCM | Mildly dilated LV, SAM, sub-valvular AvS, mild MR, mild AR | ||
| E | Female | NRAP (p.C134Sfs*12) | DCM | Biventricular dilated cardia enlargement, moderate-severe MR | EF 10%, severely depressed LV function | |
| F | Male | NRAP (p.C134Sfs*12) | DCM | Biventricular severe dilated CMP with severe MR with TR, decreased cardiac output | Not available | Hypotonia, subtle facial dysmorphia |
| G | Female | DSC2 (p.E189K) | DCM | Dilated RV, mild/moderate biventricular systolic dysfunction | Repolarization abnormalities | cMRI: dyskinesia in anterior RVOT free wall and sub tricuspid region, microaneurysm in mid inferior wall, transmural scar in mid inferior of RV (LGE) |
| H | Male | RBCK1 (p.H260Y) | DCM | Biventricular enlargement with biventricular systolic dysfunction, LV diastolic dysfunction, global hypokinesia, mild PAH | Intraventricular conduction delay, criteria for LV hypertrophy, repolarization abnormalities | CC: coronary artery disease (lesions in distal LCx and RCA) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maurer, C.; Boleti, O.; Najarzadeh Torbati, P.; Norouzi, F.; Fowler, A.N.R.; Minaee, S.; Salih, K.H.; Taherpour, M.; Birjandi, H.; Alizadeh, B.; et al. Genetic Insights from Consanguineous Cardiomyopathy Families. Genes 2023, 14, 182. https://doi.org/10.3390/genes14010182
Maurer C, Boleti O, Najarzadeh Torbati P, Norouzi F, Fowler ANR, Minaee S, Salih KH, Taherpour M, Birjandi H, Alizadeh B, et al. Genetic Insights from Consanguineous Cardiomyopathy Families. Genes. 2023; 14(1):182. https://doi.org/10.3390/genes14010182
Chicago/Turabian StyleMaurer, Constance, Olga Boleti, Paria Najarzadeh Torbati, Farzaneh Norouzi, Anna Nicole Rebekah Fowler, Shima Minaee, Khalid Hama Salih, Mehdi Taherpour, Hassan Birjandi, Behzad Alizadeh, and et al. 2023. "Genetic Insights from Consanguineous Cardiomyopathy Families" Genes 14, no. 1: 182. https://doi.org/10.3390/genes14010182
APA StyleMaurer, C., Boleti, O., Najarzadeh Torbati, P., Norouzi, F., Fowler, A. N. R., Minaee, S., Salih, K. H., Taherpour, M., Birjandi, H., Alizadeh, B., Salih, A. F., Bijari, M., Houlden, H., Pittman, A. M., Maroofian, R., Almashham, Y. H., Karimiani, E. G., Kaski, J. P., Faqeih, E. A., ... Jamshidi, Y. (2023). Genetic Insights from Consanguineous Cardiomyopathy Families. Genes, 14(1), 182. https://doi.org/10.3390/genes14010182