Molecular Characterization of Complete Genome Sequence of an Avian Coronavirus Identified in a Backyard Chicken from Tanzania

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Total RNA Extraction and Next-Generation Sequencing
2.3. Genome Sequence Assembly, Annotation and Characterization
2.4. Sanger Sequencing
3. Results
3.1. NGS and Sequence Assembly
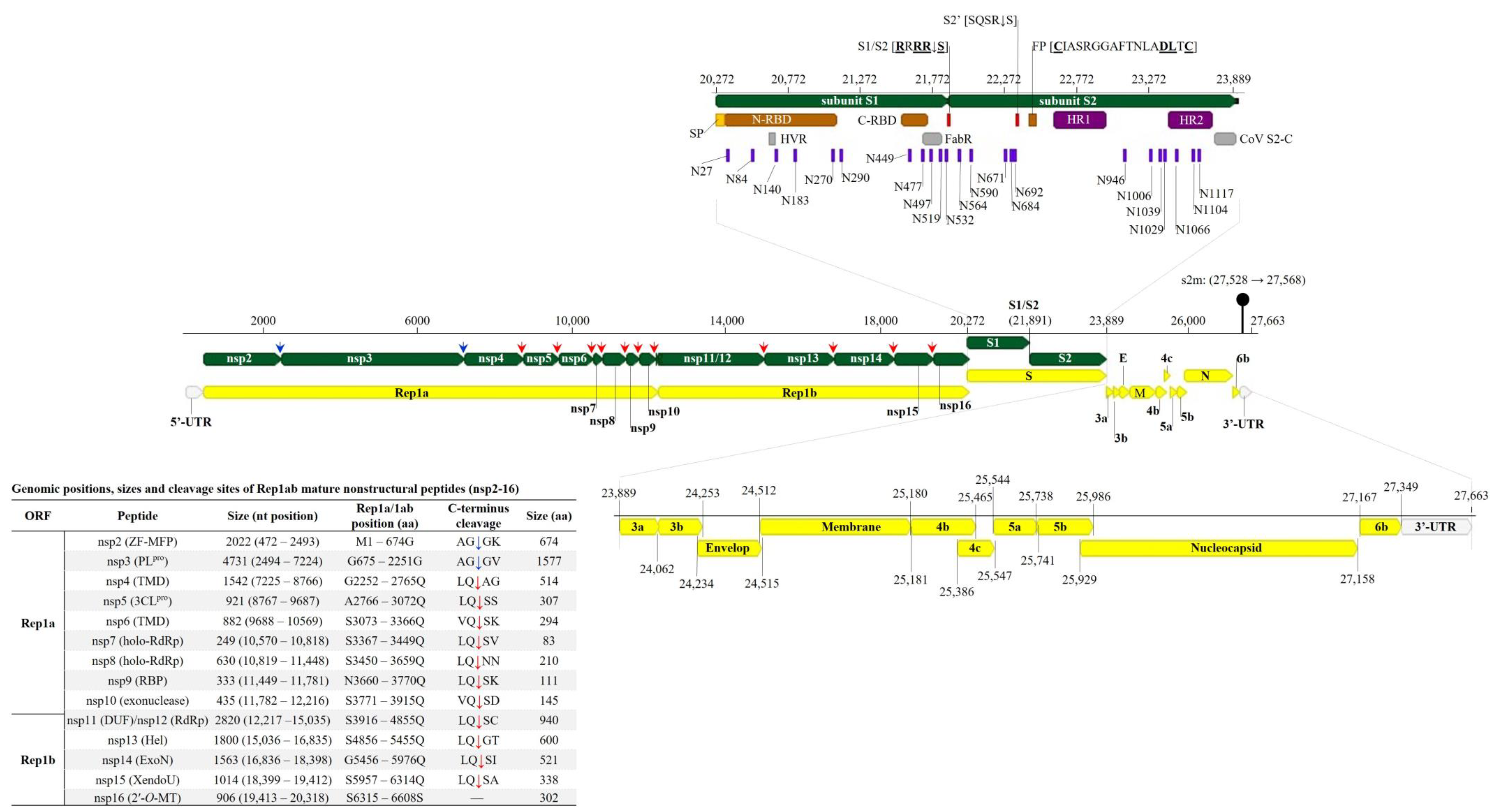
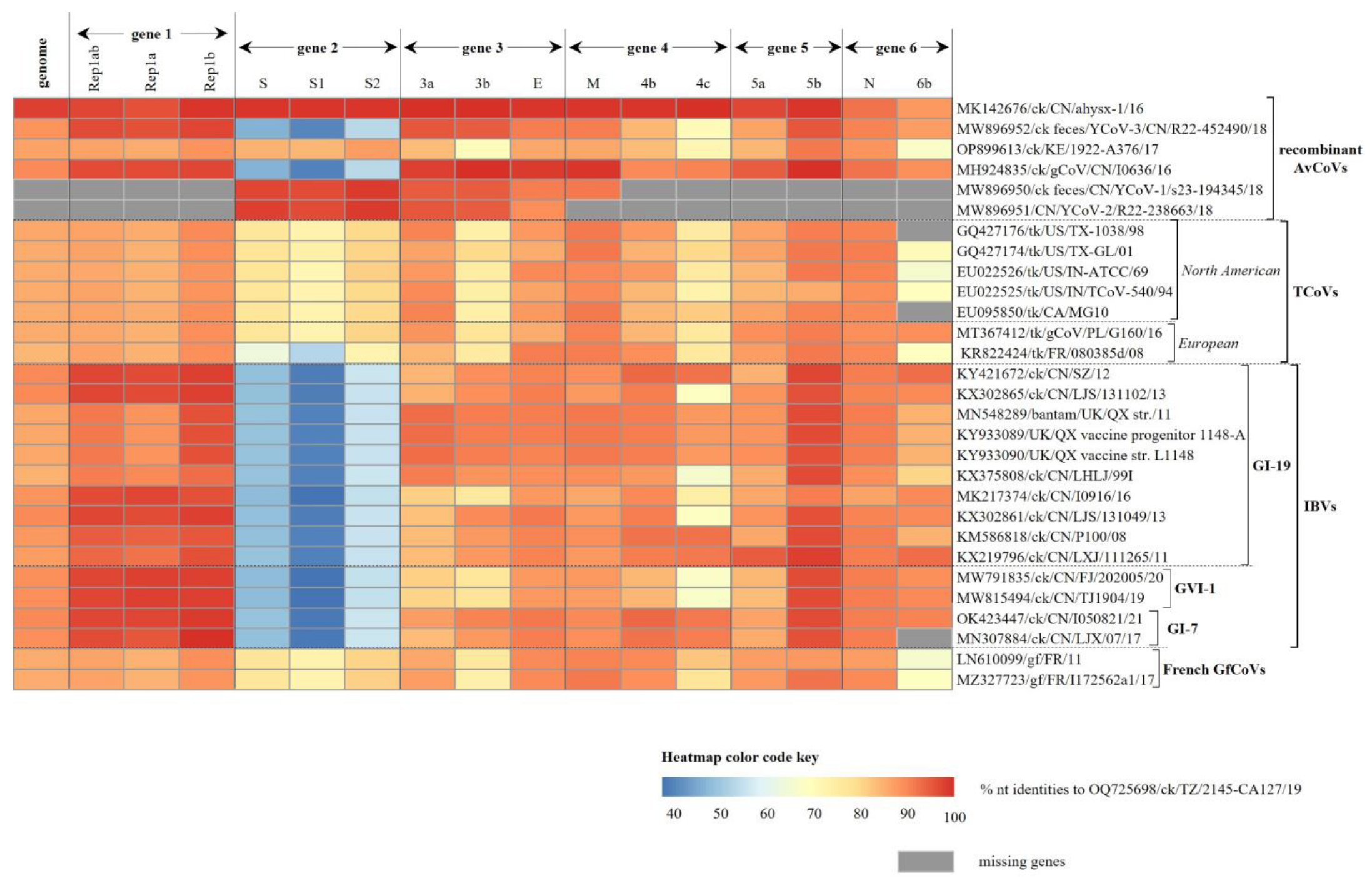
3.2. Genome Organization and Classification of the Tanzanian AvCoV
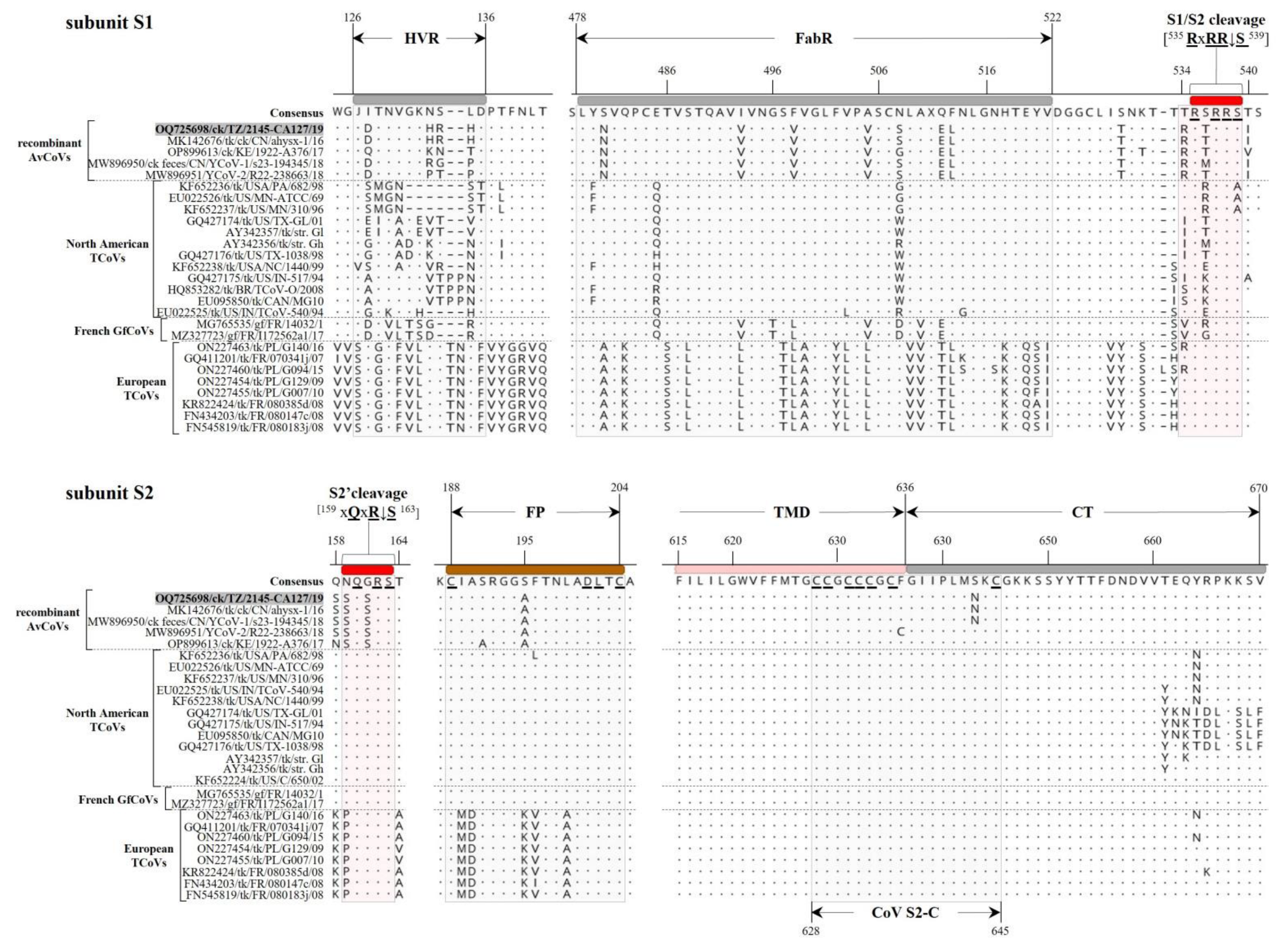
3.2.1. Structural Protein Genes
3.2.2. Non-Structural Protein Genes
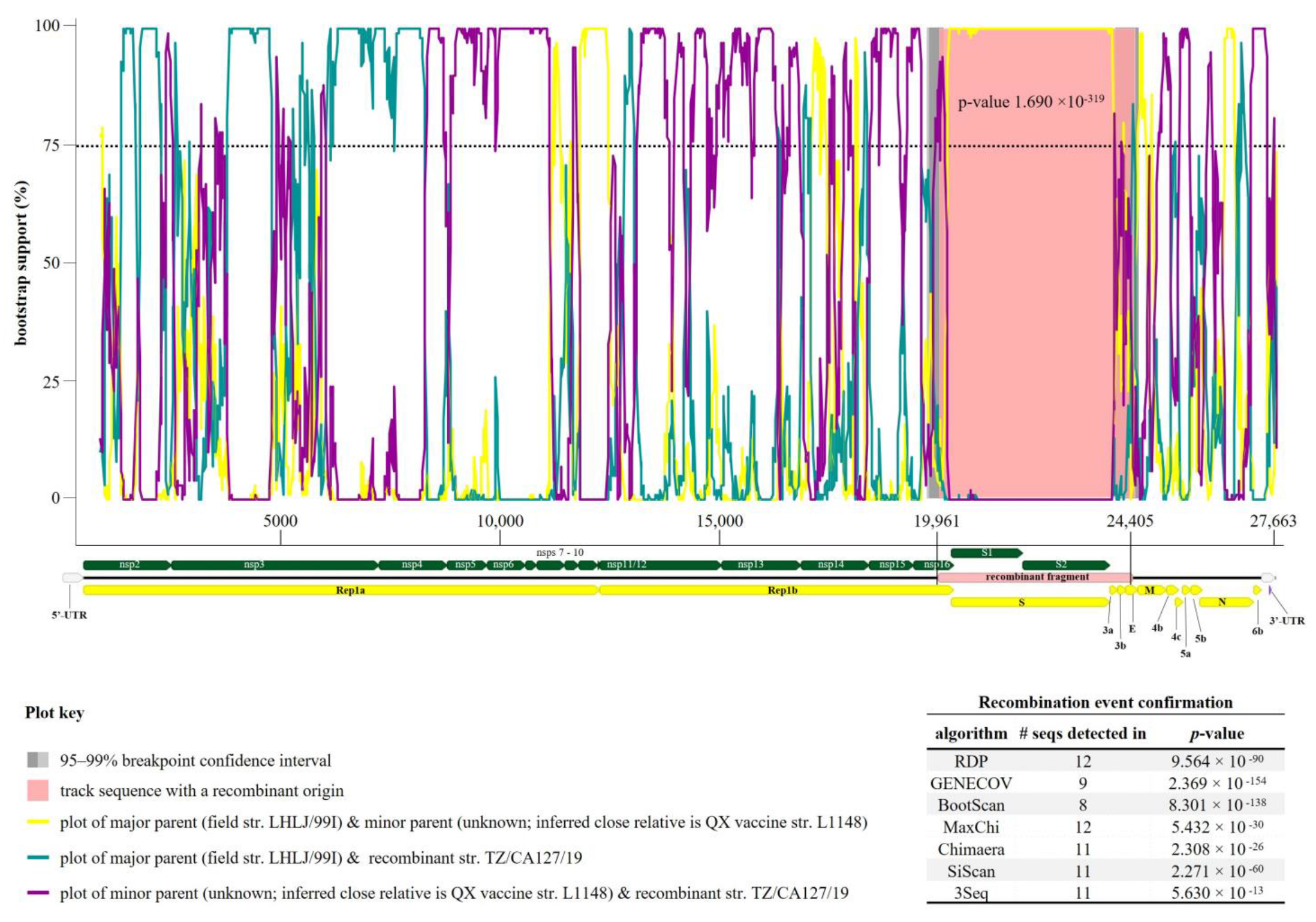
3.3. Recombination Events in Tanzanian AvCoV
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Woo, P.C.; de Groot, R.J.; Haagmans, B.; Lau, S.K.; Neuman, B.W.; Perlman, S.; Sola, I.; van der Hoek, L.; Wong, A.C.; Yeh, S.-H. ICTV Virus Taxonomy Profile: Coronaviridae 2023. J. Gen. Virol. 2023, 104, 001843. [Google Scholar] [CrossRef]
- Jackwood, M.W.; de Wit, S. CHAPTER 4: Infectious Bronchitis. In Diseases of Poultry; Swayne, D.E., Boulianne, C.M., Logue, C.M., McDougald, L.R., Nair, V., Suarez, D.L., de Wit, S., Grimes, T., Johnson, D., Kromm, M., et al., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2020; Volume 1, pp. 167–188. ISBN 978-1-119-37119-9. [Google Scholar]
- Ducatez, M.F.; European Union COST Action FA1207. Recommendations for a Standardized Avian Coronavirus (AvCoV) Nomenclature: Outcome from Discussions within the Framework of the European Union COST Action FA1207: “Towards Control of Avian Coronaviruses: Strategies for Vaccination, Diagnosis and Surveillance”. Avian Pathol. 2016, 45, 602–603. [Google Scholar] [CrossRef] [PubMed]
- (Sjaak) De Wit, J.; Cook, J.K. Spotlight on Avian Coronaviruses. Avian Pathol. 2020, 49, 313–316. [Google Scholar] [CrossRef]
- Bande, F.; Arshad, S.S.; Omar, A.R.; Hair-Bejo, M.; Mahmuda, A.; Nair, V. Global Distributions and Strain Diversity of Avian Infectious Bronchitis Virus: A Review. Anim. Health Res. Rev. 2017, 18, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Quinteros, J.; Noormohammadi, A.; Lee, S.; Browning, G.; Diaz-Méndez, A. Genomics and Pathogenesis of the Avian Coronavirus Infectious Bronchitis Virus. Aust. Vet. J. 2022, 100, 496–512. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.S. CHAPTER 12. Turkey Coronavirus Enteritis. In Diseases of Poultry; Swayne, D.E., Boulianne, M., Boulianne, C.M., McDougald, L.R., Nair, V., Suarez, D.L., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; Volume 1, pp. 402–408. ISBN 978-1-119-37116-8. [Google Scholar]
- Maurel, S.; Toquin, D.; Briand, F.-X.; Queguiner, M.; Allee, C.; Bertin, J.; Ravillion, L.; Retaux, C.; Turblin, V.; Morvan, H. First Full-Length Sequences of the S Gene of European Isolates Reveal Further Diversity among Turkey Coronaviruses. Avian Pathol. 2011, 40, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Jindal, N.; Mor, S.K.; Goyal, S.M. Enteric Viruses in Turkey Enteritis. Virus Dis. 2014, 25, 173–185. [Google Scholar] [CrossRef]
- Ziebuhr, J. CHAPTER 3. The Coronavirus Replicase. In Coronavirus Replication and Reverse Genetics; Enjuanes, L.J., Ed.; Current Topics in Microbiology and Immunology; Springer: Berlin, Heidelberg, 2005; Volume 287, pp. 57–94. ISBN 978-3-540-21494-6. [Google Scholar]
- Snijder, E.J.; Decroly, E.; Ziebuhr, J. CHAPTER 3. The Nonstructural Proteins Directing Coronavirus RNA Synthesis and Processing. In Advances in Virus Research; Ziebuhr, J., Ed.; Academic Press: Cambridge, MA, USA, 2016; Volume 96, pp. 59–126. ISBN 978-0-12-804736-1. [Google Scholar]
- Masters, P.S. The Molecular Biology of Coronaviruses. Adv. Virus Res. 2006, 66, 193–292. [Google Scholar] [CrossRef]
- Peng, S.; Wang, Y.; Zhang, Y.; Song, X.; Zou, Y.; Li, L.; Zhao, X.; Yin, Z. Current Knowledge on Infectious Bronchitis Virus Non-Structural Proteins: The Bearer for Achieving Immune Evasion Function. Front. Vet. Sci. 2022, 9, 820625. [Google Scholar] [CrossRef]
- Hewson, K.A.; Ignjatovic, J.; Browning, G.F.; Devlin, J.M.; Noormohammadi, A.H. Infectious Bronchitis Viruses with Naturally Occurring Genomic Rearrangement and Gene Deletion. Arch. Virol. 2011, 156, 245–252. [Google Scholar] [CrossRef]
- Ayres, G.R.; Brandão, P.E. Avian Coronavirus Main Replicase Enzymes at a Glance. Br. J. Virol. 2016, 3, 161–165. [Google Scholar] [CrossRef]
- Khataby, K.; Kasmi, Y.; Souiri, A.; Loutfi, C.; Ennaji, M.M. CHAPTER 33. Avian Coronavirus: Case of Infectious Bronchitis Virus Pathogenesis, Diagnostic Approaches, and Phylogenetic Relationship among Emerging Strains in Middle East and North Africa Regions. In Emerging and Reemerging Viral Pathogens; Ennaji, M.M., Ed.; Fundamental and Basic Virology Aspects of Human, Animal and Plant Pathogens; Elsevier: Amsterdam, The Netherlands, 2020; Volume 1, pp. 729–744. ISBN 978-0-12-819400-3. [Google Scholar]
- Valastro, V.; Holmes, E.C.; Britton, P.; Fusaro, A.; Jackwood, M.W.; Cattoli, G.; Monne, I. S1 Gene-Based Phylogeny of Infectious Bronchitis Virus: An Attempt to Harmonize Virus Classification. Infect. Genet. Evol. 2016, 39, 349–364. [Google Scholar] [CrossRef]
- Kariithi, H.M.; Volkening, J.D.; Goraichuk, I.V.; Ateya, L.O.; Williams-Coplin, D.; Olivier, T.L.; Binepal, Y.S.; Afonso, C.L.; Suarez, D.L. Unique Variants of Avian Coronaviruses from Indigenous Chickens in Kenya. Viruses 2023, 15, 264. [Google Scholar] [CrossRef]
- de Klerk, A.; Swanepoel, P.; Lourens, R.; Zondo, M.; Abodunran, I.; Lytras, S.; MacLean, O.A.; Robertson, D.; Kosakovsky Pond, S.L.; Zehr, J.D. Conserved Recombination Patterns across Coronavirus Subgenera. Virus Evol. 2022, 8, veac054. [Google Scholar] [CrossRef]
- Cheng, C.-P.; Nagy, P.D. Mechanism of RNA Recombination in Carmo-and Tombusviruses: Evidence for Template Switching by the RNA-Dependent RNA Polymerase in vitro. J. Virol. 2003, 77, 12033–12047. [Google Scholar] [CrossRef] [PubMed]
- Jackwood, M.W.; Boynton, T.O.; Hilt, D.A.; McKinley, E.T.; Kissinger, J.C.; Paterson, A.H.; Robertson, J.; Lemke, C.; McCall, A.W.; Williams, S.M. Emergence of a Group 3 Coronavirus through Recombination. Virology 2010, 398, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.S. Turkey Coronavirus Is More Closely Related to Avian Infectious Bronchitis Virus than to Mammalian Coronaviruses: A Review. Avian Pathol. 2000, 29, 207–212. [Google Scholar] [CrossRef]
- Wilkes, R.P.; Chan, A.; Wooming, B. Targeted Detection and Molecular Epidemiology of Turkey Coronavirus Spike Gene Variants in Turkeys and Chickens. J. Vet. Diagn. Investig. 2022, 34, 955–959. [Google Scholar] [CrossRef] [PubMed]
- Parris, D.J.; Kariithi, H.M.; Suarez, D.L. Non-Target RNA Depletion Strategy to Improve Sensitivity of next-Generation Sequencing for the Detection of RNA Viruses in Poultry. J. Vet. Diagn. Investig. 2022, 34, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, H.L.; Taylor, T.L.; Dimitrov, K.M.; Sabra, M.; Afonso, C.L.; Suarez, D.L. Virulent Newcastle Disease Viruses from Chicken Origin Are More Pathogenic and Transmissible to Chickens than Viruses Normally Maintained in Wild Birds. Vet. Microbiol. 2019, 235, 25–34. [Google Scholar] [CrossRef]
- Wise, M.G.; Suarez, D.L.; Seal, B.S.; Pedersen, J.C.; Senne, D.A.; King, D.J.; Kapczynski, D.R.; Spackman, E. Development of a Real-Time Reverse-Transcription PCR for Detection of Newcastle Disease Virus RNA in Clinical Samples. J. Clin. Microbiol. 2004, 42, 329–338. [Google Scholar] [CrossRef]
- Chrzastek, K.; Lee, D.; Smith, D.; Sharma, P.; Suarez, D.L.; Pantin-Jackwood, M.; Kapczynski, D.R. Use of Sequence-Independent, Single-Primer-Amplification (SISPA) for Rapid Detection, Identification, and Characterization of Avian RNA Viruses. Virology 2017, 509, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An Ultra-Fast Single-Node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Long-Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Kariithi, H.M.; Christy, N.; Decanini, E.L.; Lemiere, S.; Volkening, J.D.; Afonso, C.L.; Suarez, D.L. Detection and Genome Sequence Analysis of Avian Metapneumovirus Subtype A Viruses Circulating in Commercial Chicken Flocks in Mexico. Vet. Sci. 2022, 9, 579. [Google Scholar] [CrossRef]
- Kariithi, H.M.; Volkening, J.D.; Leyson, C.M.; Afonso, C.L.; Christy, N.; Decanini, E.L.; Lemiere, S.; Suarez, D.L. Genome Sequence Variations of Infectious Bronchitis Virus Serotypes from Commercial Chickens in Mexico. Front. Vet. Sci. 2022, 9, 931272. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. TrimAl: A Tool for Automated Alignment Trimming in Large-Scale Phylogenetic Analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and Analysis of Recombination Patterns in Virus Genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed]
- Metsalu, T.; Vilo, J. ClustVis: A Web Tool for Visualizing Clustering of Multivariate Data Using Principal Component Analysis and Heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]
- Keep, S.; Dowgier, G.; Lulla, V.; Britton, P.; Oade, M.; Freimanis, G.; Tennakoon, C.; Jonassen, C.M.; Tengs, T.; Bickerton, E. Deletion of the S2m RNA Structure in the Avian Coronavirus Infectious Bronchitis Virus and Human Astrovirus Results in Sequence Insertions. J. Virol. 2023, 97, e00038-23. [Google Scholar] [CrossRef]
- Keep, S.; Oade, M.S.; Lidzbarski-Silvestre, F.; Bentley, K.; Stevenson-Leggett, P.; Freimanis, G.L.; Tennakoon, C.; Sanderson, N.; Hammond, J.A.; Jones, R.C. Multiple Novel Non-Canonically Transcribed Sub-Genomic MRNAs Produced by Avian Coronavirus Infectious Bronchitis Virus. J. Gen. Virol. 2020, 101, 1103–1118. [Google Scholar] [CrossRef]
- Madu, I.G.; Roth, S.L.; Belouzard, S.; Whittaker, G.R. Characterization of a Highly Conserved Domain within the Severe Acute Respiratory Syndrome Coronavirus Spike Protein S2 Domain with Characteristics of a Viral Fusion Peptide. J. Virol. 2009, 83, 7411–7421. [Google Scholar] [CrossRef]
- Chen, Y.-N.; Wu, C.C.; Lin, T.L. Identification and Characterization of a Neutralizing-Epitope-Containing Spike Protein Fragment in Turkey Coronavirus. Arch. Virol. 2011, 156, 1525–1535. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-N.; Loa, C.C.; Ababneh, M.M.-K.; Wu, C.C.; Lin, T.L. Genotyping of Turkey Coronavirus Field Isolates from Various Geographic Locations in the Unites States Based on the Spike Gene. Arch. Virol. 2015, 160, 2719–2726. [Google Scholar] [CrossRef]
- Zhuang, Q.-Y.; Wang, K.-C.; Liu, S.; Hou, G.-Y.; Jiang, W.-M.; Wang, S.-C.; Li, J.-P.; Yu, J.-M.; Chen, J.-M. Genomic Analysis and Surveillance of the Coronavirus Dominant in Ducks in China. PLoS ONE 2015, 10, e0129256. [Google Scholar] [CrossRef] [PubMed]
- Laconi, A.; van Beurden, S.J.; Berends, A.J.; Krämer-Kühl, A.; Jansen, C.A.; Spekreijse, D.; Chénard, G.; Philipp, H.-C.; Mundt, E.; Rottier, P.J. Deletion of Accessory Genes 3a, 3b, 5a or 5b from Avian Coronavirus Infectious Bronchitis Virus Induces an Attenuated Phenotype Both in Vitro and in Vivo. J. Gen. Virol. 2018, 99, 1381. [Google Scholar] [CrossRef]
- Plant, E.P.; Dinman, J.D. The Role of Programmed-1 Ribosomal Frameshifting in Coronavirus Propagation. Front. Biosci. 2008, 13, 4873–4881. [Google Scholar] [CrossRef]
- Fang, S.; Shen, H.; Wang, J.; Tay, F.P.; Liu, D.X. Functional and Genetic Studies of the Substrate Specificity of Coronavirus Infectious Bronchitis Virus 3C-like Proteinase. J. Virol. 2010, 84, 7325–7336. [Google Scholar] [CrossRef] [PubMed]
- te Velthuis, A.J. Common and Unique Features of Viral RNA-Dependent Polymerases. Cell. Mol. Life Sci. 2014, 71, 4403–4420. [Google Scholar] [CrossRef]
- Hajijafari Anaraki, M.; Sheikhi, N.; Haghbin Nazarpak, H.; Nikbakht Brujeni, G. Molecular Characterization of Infectious Bronchitis Virus Based on RNA-dependent RNA Polymerase Gene. Microbiol. Immunol. 2020, 64, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Hemida, M.; Barta, J.; Ojkic, D.; Yoo, D. Complete Genomic Sequence of Turkey Coronavirus. Virus Res. 2008, 135, 237–246. [Google Scholar] [CrossRef]
- Domanska-Blicharz, K.; Sajewicz-Krukowska, J. Recombinant Turkey Coronavirus: Are Some S Gene Structures of Gammacoronaviruses Especially Prone to Exchange? Poult. Sci. 2021, 100, 101018. [Google Scholar] [CrossRef]
- Listorti, V.; Laconi, A.; Catelli, E.; Cecchinato, M.; Lupini, C.; Naylor, C.J. Identification of IBV QX Vaccine Markers: Should Vaccine Acceptance by Authorities Require Similar Identifications for All Live IBV Vaccines? Vaccine 2017, 35, 5531–5534. [Google Scholar] [CrossRef]
- Conan, A.; Goutard, F.L.; Sorn, S.; Vong, S. Biosecurity Measures for Backyard Poultry in Developing Countries: A Systematic Review. BMC Vet. Res. 2012, 8, 240. [Google Scholar] [CrossRef]
- Gueye, E. The Role of Networks in Information Dissemination to Family Poultry Farmers. Worlds Poult. Sci. J. 2009, 65, 115–124. [Google Scholar] [CrossRef]
- Mujyambere, V.; Adomako, K.; Olympio, S.O.; Ntawubizi, M.; Nyinawamwiza, L.; Mahoro, J.; Conroy, A. Local Chickens in East African Region: Their Production and Potential. Poult. Sci. 2022, 101, 101547. [Google Scholar] [CrossRef]
- Msoffe, P.L.; Chiwanga, G.H.; Cardona, C.J.; Miller, P.J.; Suarez, D.L. Isolation and Characterization of Newcastle Disease Virus from Live Bird Markets in Tanzania. Avian Dis. 2019, 63, 634–640. [Google Scholar] [CrossRef] [PubMed]
- (Sjaak) De Wit, J.; Cook, J.K.; van der Heijden, H.M. Infectious Bronchitis Virus Variants: A Review of the History, Current Situation and Control Measures. Avian Pathol. 2011, 40, 223–235. [Google Scholar] [CrossRef]
- Cook, J.K.; Jackwood, M.; Jones, R. The Long View: 40 Years of Infectious Bronchitis Research. Avian Pathol. 2012, 41, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Bali, K.; Kaszab, E.; Marton, S.; Hamdiou, S.H.; Bentaleb, R.K.; Kiss, I.; Palya, V.; Bányai, K. Novel Lineage of Infectious Bronchitis Virus from Sub-Saharan Africa Identified by Random Amplification and next-Generation Sequencing of Viral Genome. Life 2022, 12, 475. [Google Scholar] [CrossRef] [PubMed]
- Alders, R.; Bagnol, B.; Young, M. Technically Sound and Sustainable Newcastle Disease Control in Village Chickens: Lessons Learnt over Fifteen Years. Worlds Poult. Sci. J. 2010, 66, 433–440. [Google Scholar] [CrossRef]
- Britton, P.; Casais, R.; Hodgson, T.; Davis, M.; Cavanagh, D. Genes 3 and 5 of Infectious Bronchitis Virus Are Accessory Protein Genes. Adv. Exp. Med. Biol. 2006, 581, 363–368. [Google Scholar] [CrossRef]
- Bentley, K.; Keep, S.M.; Armesto, M.; Britton, P. Identification of a Noncanonically Transcribed Subgenomic MRNA of Infectious Bronchitis Virus and Other Gammacoronaviruses. J. Virol. 2013, 87, 2128–2136. [Google Scholar] [CrossRef] [PubMed]
- Casais, R.; Davies, M.; Cavanagh, D.; Britton, P. Gene 5 of the Avian Coronavirus Infectious Bronchitis Virus Is Not Essential for Replication. J. Virol. 2005, 79, 8065–8078. [Google Scholar] [CrossRef]
- Yang, Y.; Yan, W.; Hall, A.B.; Jiang, X. Characterizing Transcriptional Regulatory Sequences in Coronaviruses and Their Role in Recombination. Mol. Biol. Evol. 2021, 38, 1241–1248. [Google Scholar] [CrossRef]
- Franzo, G.; Naylor, C.J.; Lupini, C.; Drigo, M.; Catelli, E.; Listorti, V.; Pesente, P.; Giovanardi, D.; Morandini, E.; Cecchinato, M. Continued Use of IBV 793B Vaccine Needs Reassessment after Its Withdrawal Led to the Genotype’s Disappearance. Vaccine 2014, 32, 6765–6767. [Google Scholar] [CrossRef]
- Franzo, G.; Massi, P.; Tucciarone, C.M.; Barbieri, I.; Tosi, G.; Fiorentini, L.; Ciccozzi, M.; Lavazza, A.; Cecchinato, M.; Moreno, A. Think Globally, Act Locally: Phylodynamic Reconstruction of Infectious Bronchitis Virus (IBV) QX Genotype (GI-19 Lineage) Reveals Different Population Dynamics and Spreading Patterns When Evaluated on Different Epidemiological Scales. PLoS ONE 2017, 12, e0184401. [Google Scholar] [CrossRef]
- Li, H.; Liu, G.; Zhou, Q.; Yang, H.; Zhou, C.; Kong, W.; Su, J.; Li, G.; Si, H.; Ou, C. Which Strain of the Avian Coronavirus Vaccine Will Become the Prevalent One in China Next? Front. Vet. Sci. 2023, 10, 1139089. [Google Scholar] [CrossRef] [PubMed]
- Lian, J.; Wang, Z.; Xu, Z.; Chen, T.; Shao, G.; Zhang, X.; Qin, J.; Xie, Q.; Lin, W. Distribution and Molecular Characterization of Avian Infectious Bronchitis Virus in Southern China. Poult. Sci. 2021, 100, 101169. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Zhang, L.; Hou, Y.; Zhao, Y.; Han, Z.; Sun, J.; Liu, S. Genetic, Antigenic, and Pathogenic Characteristics of Infectious Bronchitis Virus GI-7/TW-II in China. Avian Dis. 2020, 64, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Liu, X.; Zhao, Y.; Chen, Y.; Zhao, J.; Zhang, G. Characterization and Analysis of an Infectious Bronchitis Virus Strain Isolated from Southern China in 2013. Virol. J. 2016, 13, 40. [Google Scholar] [CrossRef]
- Lin, T.L.; Loa, C.C.; Wu, C.C. Complete Sequences of 3′ End Coding Region for Structural Protein Genes of Turkey Coronavirus. Virus Res. 2004, 106, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Junker, D.; Collisson, E.W. Evidence of Natural Recombination within the S1 Gens of Infectious Bronchitis Virus. Virology 1993, 192, 710–716. [Google Scholar] [CrossRef]
- Pearson, D.J.; Lack, P.C. Migration Patterns and Habitat Use by Passerine and Near-passerine Migrant Birds in Eastern Africa. Ibis 1992, 134, 89–98. [Google Scholar] [CrossRef]
- Bande, F.; Arshad, S.S.; Hair Bejo, M.; Moeini, H.; Omar, A.R. Progress and Challenges toward the Development of Vaccines against Avian Infectious Bronchitis. J. Immunol. Res. 2015, 2015, 424860. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NGS Read Pairs | De novo Genome Assembly | Number of Missing Bases | Other Microbial Pathogens of Avian Interest Detected with High Confidence (Specific Read Count) * | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Total | Host-Specific (%) | IBV-Specific | Median Coverage Depth (Reads) | Consensus Seq. (Contig) Length | Completeness (%) | 5′-End | # Internal Gaps (Length; Position) | 3′-end | Viral (Specific Reads) | Bacterial (Specific Reads) |
| 510,552 | 8.41% | 26,485 | 262 | 27,663 | 98.50% | 42 | 8-nt; 11,064–11,071 | 0 | vNDV-VII.2 (177,140); ChMeV (110); AAstV (101) | E. faecium (202); ORT (1638); R. anatipestifer (1143); M. synoviae (n = 953); A. paragallinarum (303) |
| Gene | Reading Frame | Gene Product | Genomic Position | Nucleotide Length (aa) | Putative Transcription Regulatory Sequence (TRS) | ||
|---|---|---|---|---|---|---|---|
| Sequence a | Position | TRS—Initiation Distance (nt) b | |||||
| 5’-UTR | - | - | 1–471 | 471 | - | - | - |
| Gene 1 | +1 | Rep1a | 472–12,288 | 11,817 (3938) | CTTAACAA | 3–10 | 461 |
| Rep1ab | 472–20,321 | 19,851 (6616) | |||||
| Gene 2 | +1 | S1 | 20,272–21,891 | 1620 (540) | CTGAACAA | 20,212–20,219 | 52 |
| S2 | 21,892–23,889 | 1998 (665) | |||||
| Gene 3 | +3 | 3a | 23,889–24,062 | 174 (57) | CTGAACAA | 23,858–23,865 | 23 |
| +2 | 3b | 24,062–24,253 | 192 (63) | ||||
| +3 | E | 24,234–24,515 | 282 (93) | ||||
| Gene 4 | +2 | M | 24,512–25,180 | 669 (222) | CTTAACAA | 24,450–24,457 | 54 |
| +2 | 4b | 25,181–25,465 | 285 (94) | ||||
| +3 | 4c | 25,386–25,547 | 162 (53) | ||||
| Gene 5 | +2 | 5a | 25,544–25,741 | 198 (65) | AACAACAA | 25,481–25,488 | 55 |
| +1 | 5b | 25,738–25,986 | 249 (82) | ||||
| Gene 6 | +3 | N | 25,929–27,158 | 1230 (409) | CTTAACAA | 25,828–25,835 | 93 |
| +2 | 6b | 27,167–27,349 | 183 (60) | AGGAACAA | 27,075–27,082 | 84 | |
| 3’-UTR | - | - | 27,350–27,663 | 314 | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kariithi, H.M.; Volkening, J.D.; Chiwanga, G.H.; Goraichuk, I.V.; Msoffe, P.L.M.; Suarez, D.L. Molecular Characterization of Complete Genome Sequence of an Avian Coronavirus Identified in a Backyard Chicken from Tanzania. Genes 2023, 14, 1852. https://doi.org/10.3390/genes14101852
Kariithi HM, Volkening JD, Chiwanga GH, Goraichuk IV, Msoffe PLM, Suarez DL. Molecular Characterization of Complete Genome Sequence of an Avian Coronavirus Identified in a Backyard Chicken from Tanzania. Genes. 2023; 14(10):1852. https://doi.org/10.3390/genes14101852
Chicago/Turabian StyleKariithi, Henry M., Jeremy D. Volkening, Gaspar H. Chiwanga, Iryna V. Goraichuk, Peter L. M. Msoffe, and David L. Suarez. 2023. "Molecular Characterization of Complete Genome Sequence of an Avian Coronavirus Identified in a Backyard Chicken from Tanzania" Genes 14, no. 10: 1852. https://doi.org/10.3390/genes14101852