
GIP_HUMAN [22–51] Peptide Encoded by the Glucose-Dependent Insulinotropic Polypeptide (GIP) Gene Suppresses Insulin Expression and Secretion in INS-1E Cells and Rat Pancreatic Islets
 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Isolation of Pancreatic Islets
2.4. Real-Time PCR
2.5. Immunofluorescence Detection of NF-κB Nuclear Translocation
2.6. Insulin Secretion
2.7. Determination of Intracellular Insulin Content
2.8. Cell Proliferation
2.9. Cell Death
2.10. Statistical Analysis
3. Results
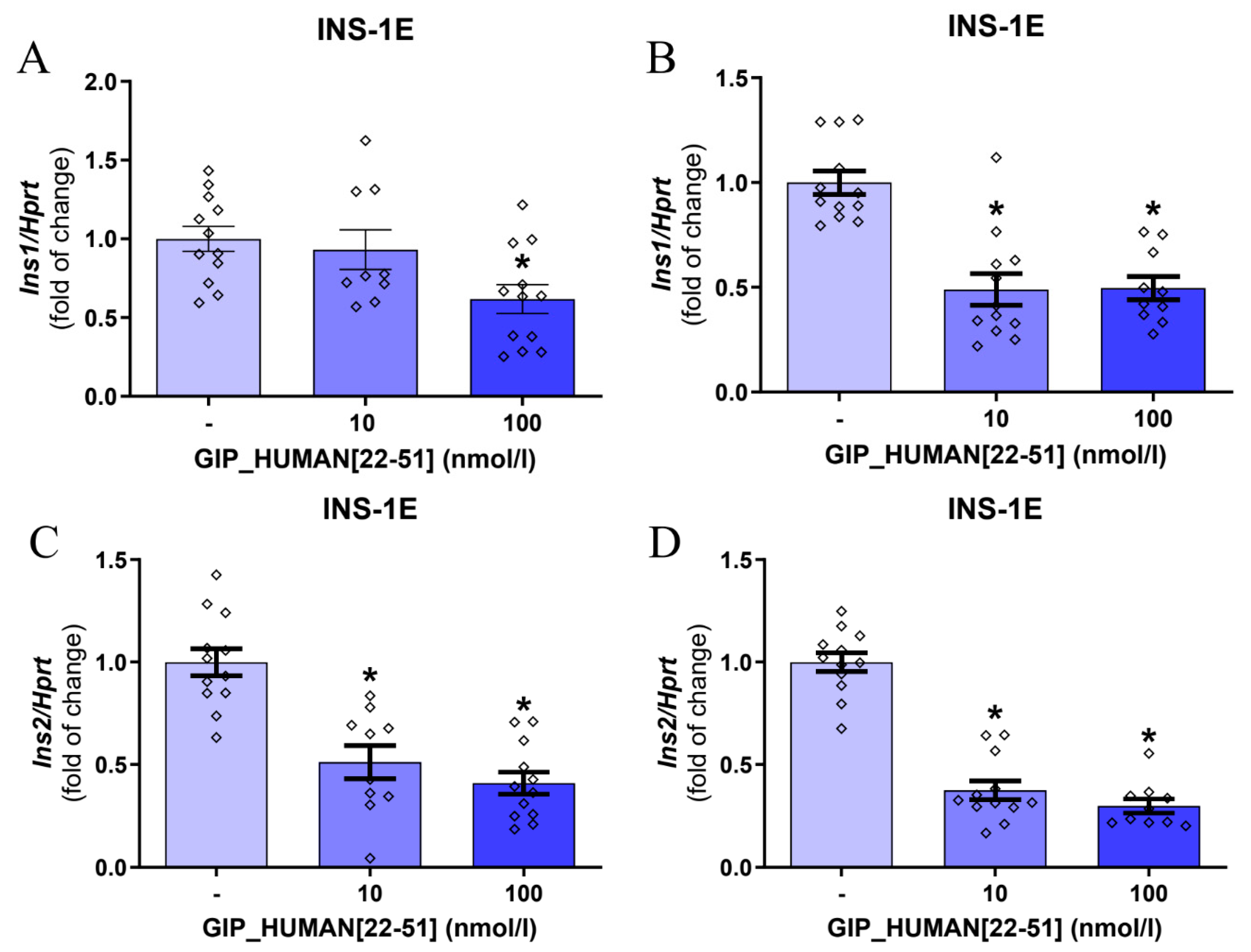
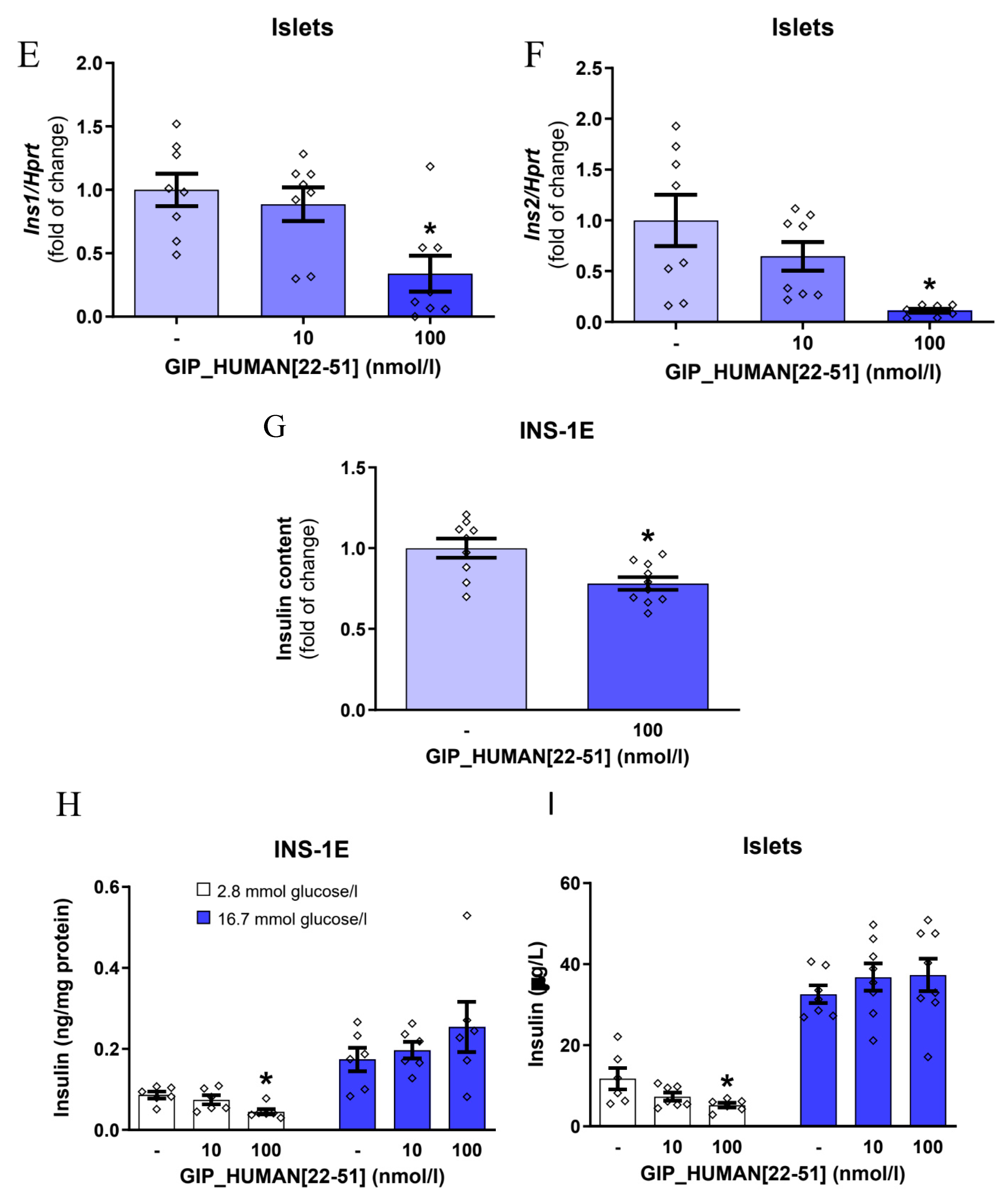
3.1. GIP_HUMAN [22–51] Modulates Insulin mRNA in INS-1E Cells and Isolated Rat Pancreatic Islets
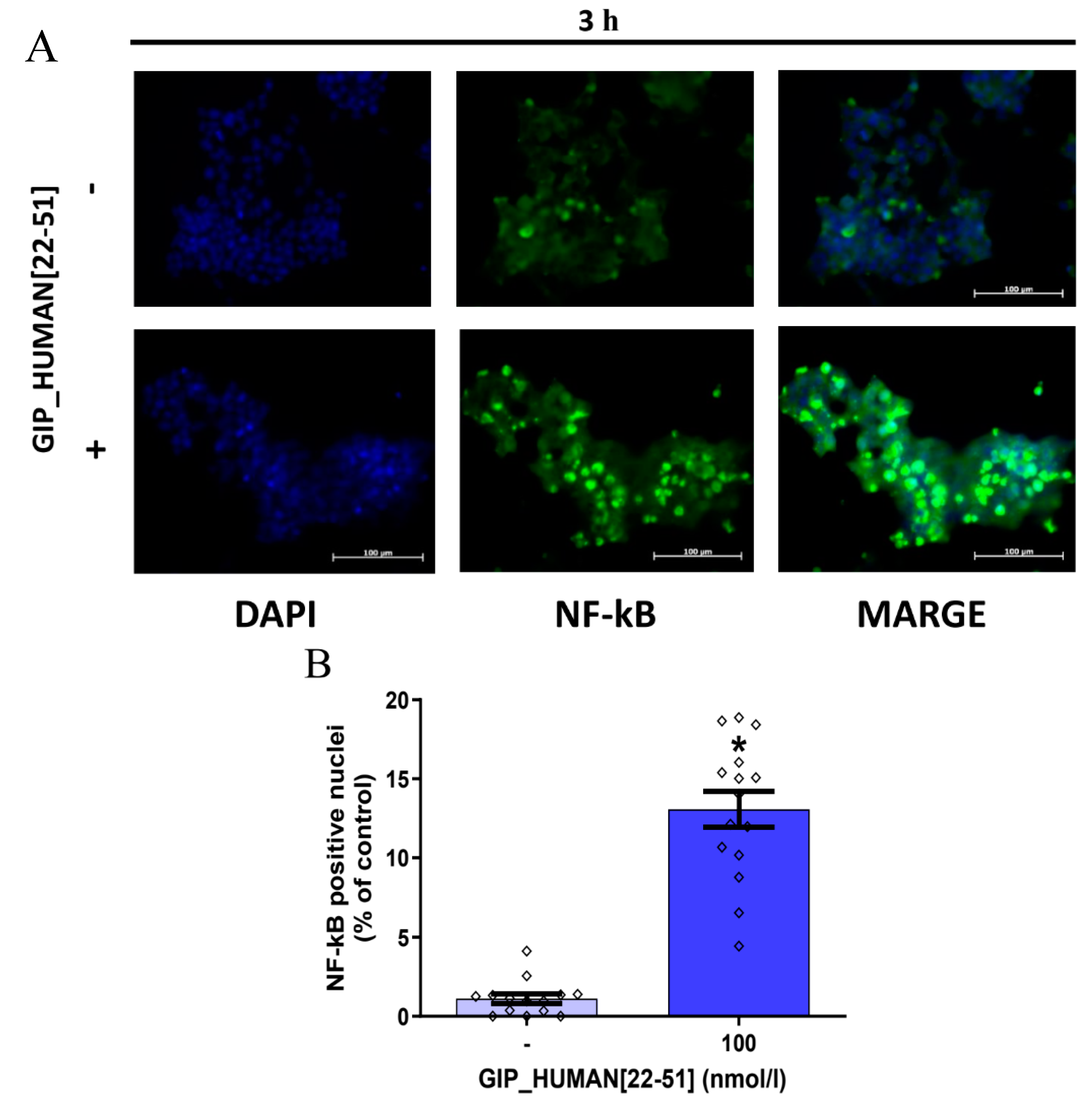
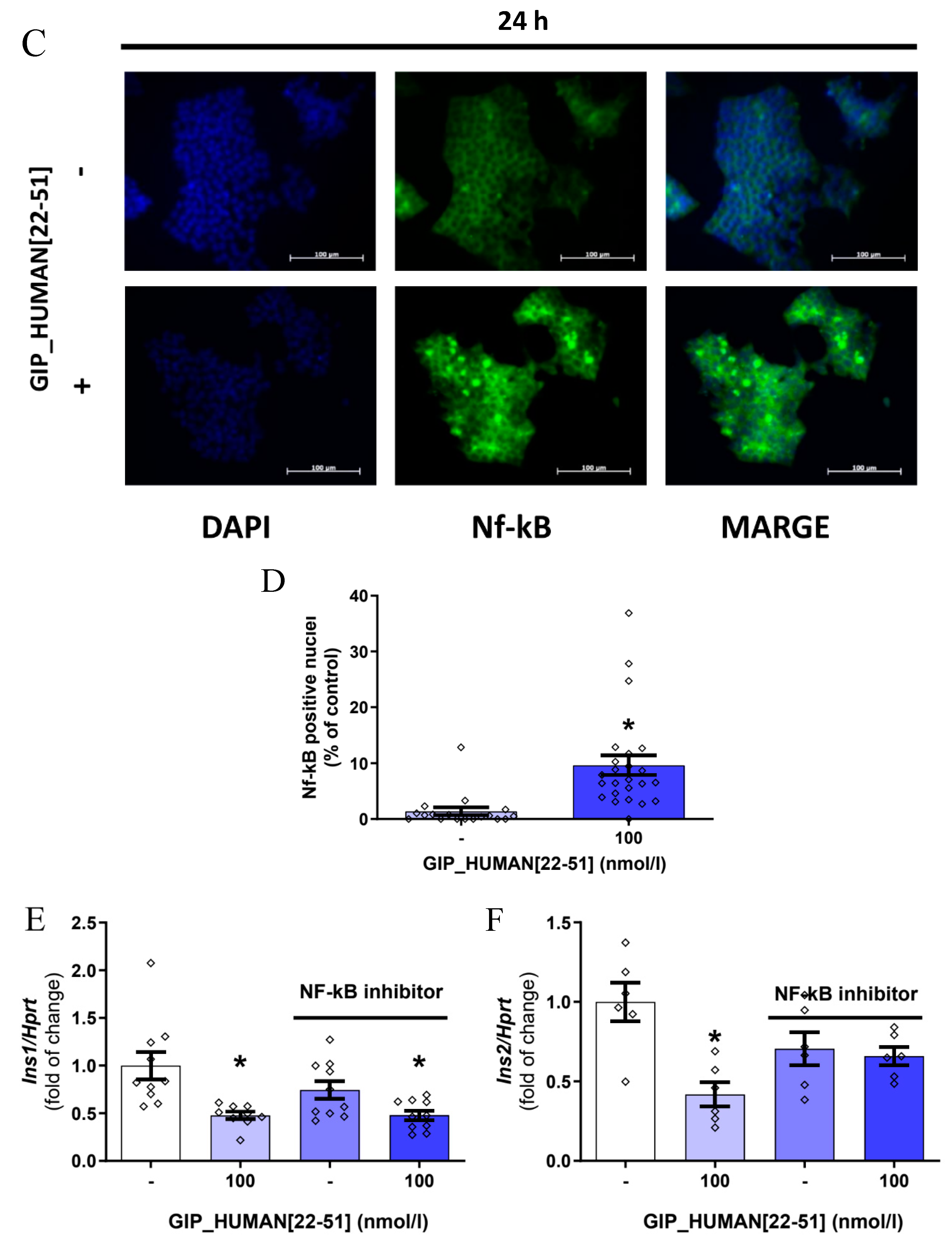
3.2. GIP_HUMAN [22–51] Activates NF-kB in INS-1E Cells
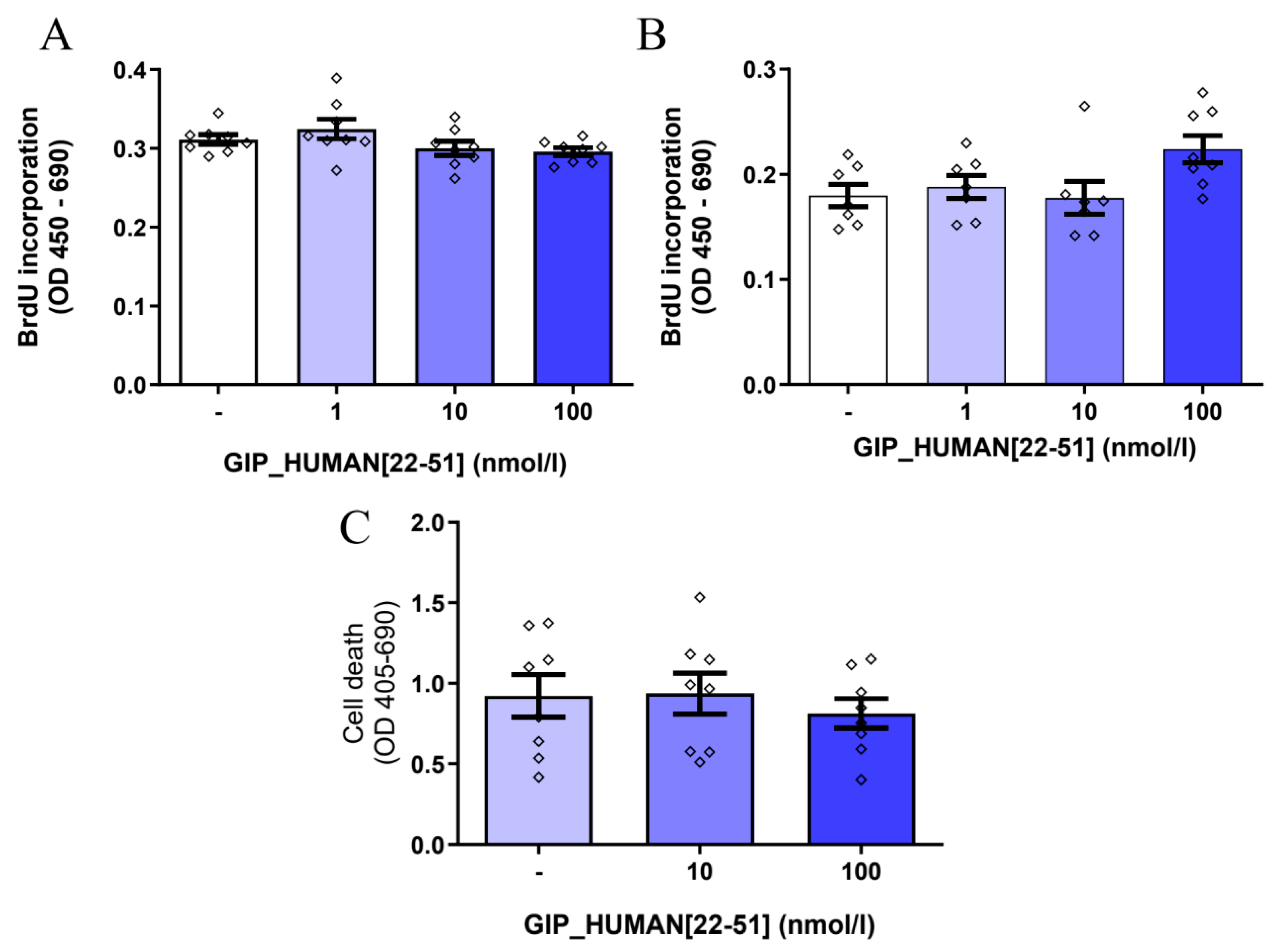
3.3. GIP_HUMAN [22–51] Does Not Modulate Cell Proliferation and Death in INS-1E Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aronoff, S.L.; Berkowitz, K.; Shreiner, B.; Want, L. Glucose Metabolism and Regulation: Beyond Insulin and Glucagon. Diabetes Spectr. 2004, 17, 183–190. [Google Scholar] [CrossRef]
- Katsarou, A.; Gudbjörnsdottir, S.; Rawshani, A.; Dabelea, D.; Bonifacio, E.; Anderson, B.J.; Jacobsen, L.M.; Schatz, D.A.; Lernmark, Å. Type 1 Diabetes Mellitus. Nat. Rev. Dis. Primers 2017, 3, 17016. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Ferrannini, E.; Groop, L.; Henry, R.R.; Herman, W.H.; Holst, J.J.; Hu, F.B.; Kahn, C.R.; Raz, I.; Shulman, G.I.; et al. Type 2 Diabetes Mellitus. Nat. Rev. Dis. Primers 2015, 1, 15019. [Google Scholar] [CrossRef]
- Seino, Y.; Fukushima, M.; Yabe, D. GIP and GLP-1, the Two Incretin Hormones: Similarities and Differences. J. Diabetes Investig. 2010, 1, 8–23. [Google Scholar] [CrossRef]
- MacDonald, P.E.; El-kholy, W.; Riedel, M.J.; Salapatek, A.M.F.; Light, P.E.; Wheeler, M.B. The Multiple Actions of GLP-1 on the Process of Glucose-Stimulated Insulin Secretion. Diabetes 2002, 51, S434–S442. [Google Scholar] [CrossRef]
- Mayendraraj, A.; Rosenkilde, M.M.; Gasbjerg, L.S. GLP-1 and GIP Receptor Signaling in Beta Cells—A Review of Receptor Interactions and Co-Stimulation. Peptides 2022, 151, 170749. [Google Scholar] [CrossRef]
- Holst, J.J.; Jepsen, S.L.; Modvig, I. GLP-1—Incretin and Pleiotropic Hormone with Pharmacotherapy Potential. Increasing Secretion of Endogenous GLP-1 for Diabetes and Obesity Therapy. Curr. Opin. Pharmacol. 2022, 63, 102189. [Google Scholar] [CrossRef]
- Masaki, T.; Kodera, Y.; Terasaki, M.; Fujimoto, K.; Hirano, T.; Shichiri, M. GIP_HUMAN [22–51] Is a New Proatherogenic Peptide Identified by Native Plasma Peptidomics. Sci. Rep. 2021, 11, 14470. [Google Scholar] [CrossRef]
- van Diepen, J.A.; Thiem, K.; Stienstra, R.; Riksen, N.P.; Tack, C.J.; Netea, M.G. Diabetes Propels the Risk for Cardiovascular Disease: Sweet Monocytes Becoming Aggressive? Cell. Mol. Life Sci. 2016, 73, 4675–4684. [Google Scholar] [CrossRef]
- Juutilainen, A.; Lehto, S.; Rönnemaa, T.; Pöyrälä, K.; Laakso, M. Similarity of the Impact of Type 1 and Type 2 Diabetes on Cardiovascular Mortality in Middle-Aged Subjects. Diabetes Care 2008, 31, 714–719. [Google Scholar] [CrossRef]
- Maechler, P.; Antinozzi, P.A.; Wollheim, C.B. Modulation of Glutamate Generation in Mitochondria Affects Hormone Secretion in INS-1E Beta Cells. IUBMB Life 2000, 50, 27–31. [Google Scholar] [CrossRef]
- Billert, M.; Jasaszwili, M.; Strowski, M.; Nowak, K.W.; Skrzypski, M. Adropin Suppresses Insulin Expression and Secretion in INS-1E Cells and Rat Pancreatic Islets. J. Physiol. Pharmacol. 2020, 71, 99–104. [Google Scholar] [CrossRef]
- Andreone, L.; Fuertes, F.; Sétula, C.; Barcala Tabarrozzi, A.E.; Orellano, M.S.; Dewey, R.A.; Bottino, R.; De Bosscher, K.; Perone, M.J. Compound A Attenuates Proinflammatory Cytokine-Induced Endoplasmic Reticulum Stress in Beta Cells and Displays Beneficial Therapeutic Effects in a Mouse Model of Autoimmune Diabetes. Cell. Mol. Life Sci. 2022, 79, 587. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Tian, W.; Pratt, E.B.; Dirling, L.B.; Shyng, S.-L.; Meshul, C.K.; Cohen, D.M. Down-Regulation of ZnT8 Expression in INS-1 Rat Pancreatic Beta Cells Reduces Insulin Content and Glucose-Inducible Insulin Secretion. PLoS ONE 2009, 4, e5679. [Google Scholar] [CrossRef] [PubMed]
- Paronen, J.; Moriyama, H.; Abiru, N.; Sikora, K.; Melanitou, E.; Babu, S.; Bao, F.; Liu, E.; Miao, D.; Eisenbarth, G.S. Establishing Insulin 1 and Insulin 2 Knockout Congenic Strains on NOD Genetic Background. Ann. N.Y. Acad. Sci. 2003, 1005, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Gilbert, E.R.; Liu, D. Regulation of Insulin Synthesis and Secretion and Pancreatic Beta-Cell Dysfunction in Diabetes. Curr. Diabetes Rev. 2013, 9, 25–53. [Google Scholar] [CrossRef] [PubMed]
- Blum, B.; Hrvatin, S.; Schuetz, C.; Bonal, C.; Rezania, A.; Melton, D.A. Functional Beta-Cell Maturation Is Marked by an Increased Glucose Threshold and by Expression of Urocortin 3. Nat. Biotechnol. 2012, 30, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Amyot, J.; Semache, M.; Ferdaoussi, M.; Fontés, G.; Poitout, V. Lipopolysaccharides Impair Insulin Gene Expression in Isolated Islets of Langerhans via Toll-like Receptor-4 and NF-ΚB Signalling. PLoS ONE 2012, 7, e36200. [Google Scholar] [CrossRef]
- Eldor, R.; Yeffet, A.; Baum, K.; Doviner, V.; Amar, D.; Ben-Neriah, Y.; Christofori, G.; Peled, A.; Carel, J.C.; Boitard, C.; et al. Conditional and Specific NF-KappaB Blockade Protects Pancreatic Beta Cells from Diabetogenic Agents. Proc. Natl. Acad. Sci. USA 2006, 103, 5072–5077. [Google Scholar] [CrossRef]
- Meyerovich, K.; Ortis, F.; Cardozo, A.K. The Non-Canonical NF-ΚB Pathway and Its Contribution to β-Cell Failure in Diabetes. J. Mol. Endocrinol. 2018, 61, F1–F6. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-ΚB, Inflammation, Immunity and Cancer: Coming of Age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Heynekamp, J.J.; Weber, W.M.; Hunsaker, L.A.; Gonzales, A.M.; Orlando, R.A.; Deck, L.M.; Jagt, D.L. Vander Substituted Trans-Stilbenes, Including Analogues of the Natural Product Resveratrol, Inhibit the Human Tumor Necrosis Factor Alpha-Induced Activation of Transcription Factor Nuclear Factor KappaB. J. Med. Chem. 2006, 49, 7182–7189. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Stein, G.H.; Pan, N.; Goebbels, S.; Hörnberg, H.; Nave, K.-A.; Herrera, P.; White, P.; Kaestner, K.H.; Sussel, L.; et al. Pancreatic Beta Cells Require NeuroD to Achieve and Maintain Functional Maturity. Cell Metab. 2010, 11, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Kiba, T. Overexpression of PDX-1 Gene Increases INS1 Gene MRNA Expression, not INS2 Gene MRNA Expression, In Insulinoma Cell Line RIN-5F. Acta Endocrinol. 2022, 18, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, Q.; Zhou, Z.; Ikeda, Y. PDX1, Neurogenin-3, and MAFA: Critical Transcription Regulators for Beta Cell Development and Regeneration. Stem Cell Res. Ther. 2017, 8, 240. [Google Scholar] [CrossRef] [PubMed]
- Bataille, D. Molecular Mechanisms of Insulin Secretion. Diabetes Metab. 2002, 28, S7–S13. [Google Scholar]
- Corkey, B.E.; Deeney, J.T.; Merrins, M.J. What Regulates Basal Insulin Secretion and Causes Hyperinsulinemia? Diabetes 2021, 70, 2174–2182. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, V.; Kazim, A.S.; Helgeson, J.; Lewold, C.; Barik, S.; Buda, P.; Reinbothe, T.M.; Wennmalm, S.; Zhang, E.; Renström, E. Elevated Basal Insulin Secretion in Type 2 Diabetes Caused by Reduced Plasma Membrane Cholesterol. Mol. Endocrinol. 2016, 30, 1059–1069. [Google Scholar] [CrossRef]
- Philippaert, K.; Vennekens, R. The Role of TRP Channels in the Pancreatic Beta-Cell. In Neurobiology of TRP Channels; CRC Press: Boca Raton, FL, USA, 2017; pp. 229–250. [Google Scholar]
- Skrzypski, M.; Billert, M.; Mergler, S.; Khajavi, N.; Nowak, K.W.; Strowski, M.Z. Role of TRPV Channels in Regulating Various Pancreatic β-Cell Functions: Lessons from in Vitro Studies. Biosci. Trends 2017, 11, 9–15. [Google Scholar] [CrossRef]
- Manialawy, Y.; Khan, S.R.; Bhattacharjee, A.; Wheeler, M.B. The Magnesium Transporter NIPAL1 Is a Pancreatic Islet–Expressed Protein That Conditionally Impacts Insulin Secretion. J. Biol. Chem. 2020, 295, 9879–9892. [Google Scholar] [CrossRef]
- Li, X.; Wu, Y.; Song, Y.; Ding, N.; Lu, M.; Jia, L.; Zhao, Y.; Liu, M.; Chen, Z. Activation of NF-ΚB-Inducing Kinase in Islet β Cells Causes β Cell Failure and Diabetes. Mol. Ther. 2020, 28, 2430–2441. [Google Scholar] [CrossRef] [PubMed]
- Sever, D.; Hershko-Moshe, A.; Srivastava, R.; Eldor, R.; Hibsher, D.; Keren-Shaul, H.; Amit, I.; Bertuzzi, F.; Krogvold, L.; Dahl-Jørgensen, K.; et al. NF-ΚB Activity during Pancreas Development Regulates Adult β-Cell Mass by Modulating Neonatal β-Cell Proliferation and Apoptosis. Cell Death Discov. 2021, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Millet, I.; Kim, H.S.; Kim, J.Y.; Han, M.S.; Lee, M.-K.; Kim, K.-W.; Sherwin, R.S.; Karin, M.; Lee, M.-S. NF-Kappa B Prevents Beta Cell Death and Autoimmune Diabetes in NOD Mice. Proc. Natl. Acad. Sci. USA 2007, 104, 1913–1918. [Google Scholar] [CrossRef] [PubMed]
- Chang, I.; Kim, S.; Kim, J.Y.; Cho, N.; Kim, Y.-H.; Kim, H.S.; Lee, M.-K.; Kim, K.-W.; Lee, M.-S. Nuclear Factor KappaB Protects Pancreatic Beta-Cells from Tumor Necrosis Factor-Alpha-Mediated Apoptosis. Diabetes 2003, 52, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-ΚB Pathway for the Therapy of Diseases: Mechanism and Clinical Study. Signal Transduct. Target. Ther. 2020, 5, 209. [Google Scholar] [CrossRef] [PubMed]
- Israel, A. The IKK Complex, a Central Regulator of NF- B Activation. Cold Spring Harb. Perspect. Biol. 2010, 2, a000158. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Li, L.; Wang, M.; Ma, Q.; Tian, Y.; Zhang, Q.; Liu, J.; Li, B.; Zhang, B.; Liu, H.; et al. Diabetes Mellitus Promotes the Development of Atherosclerosis: The Role of NLRP3. Front. Immunol. 2022, 13, 900254. [Google Scholar] [CrossRef] [PubMed]
- Ahren, B. Type 2 Diabetes, Insulin Secretion and β-Cell Mass. Curr. Mol. Med. 2005, 5, 275–286. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pusch, E.; Krążek, M.; Wojciechowicz, T.; Sassek, M.; Kołodziejski, P.A.; Strowski, M.Z.; Nowak, K.W.; Skrzypski, M. GIP_HUMAN [22–51] Peptide Encoded by the Glucose-Dependent Insulinotropic Polypeptide (GIP) Gene Suppresses Insulin Expression and Secretion in INS-1E Cells and Rat Pancreatic Islets. Genes 2023, 14, 1910. https://doi.org/10.3390/genes14101910
Pusch E, Krążek M, Wojciechowicz T, Sassek M, Kołodziejski PA, Strowski MZ, Nowak KW, Skrzypski M. GIP_HUMAN [22–51] Peptide Encoded by the Glucose-Dependent Insulinotropic Polypeptide (GIP) Gene Suppresses Insulin Expression and Secretion in INS-1E Cells and Rat Pancreatic Islets. Genes. 2023; 14(10):1910. https://doi.org/10.3390/genes14101910
Chicago/Turabian StylePusch, Emily, Małgorzata Krążek, Tatiana Wojciechowicz, Maciej Sassek, Paweł A. Kołodziejski, Mathias Z. Strowski, Krzysztof W. Nowak, and Marek Skrzypski. 2023. "GIP_HUMAN [22–51] Peptide Encoded by the Glucose-Dependent Insulinotropic Polypeptide (GIP) Gene Suppresses Insulin Expression and Secretion in INS-1E Cells and Rat Pancreatic Islets" Genes 14, no. 10: 1910. https://doi.org/10.3390/genes14101910