
Genome-Wide Analysis on Driver and Passenger RNA Editing Sites Suggests an Underestimation of Adaptive Signals in Insects


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
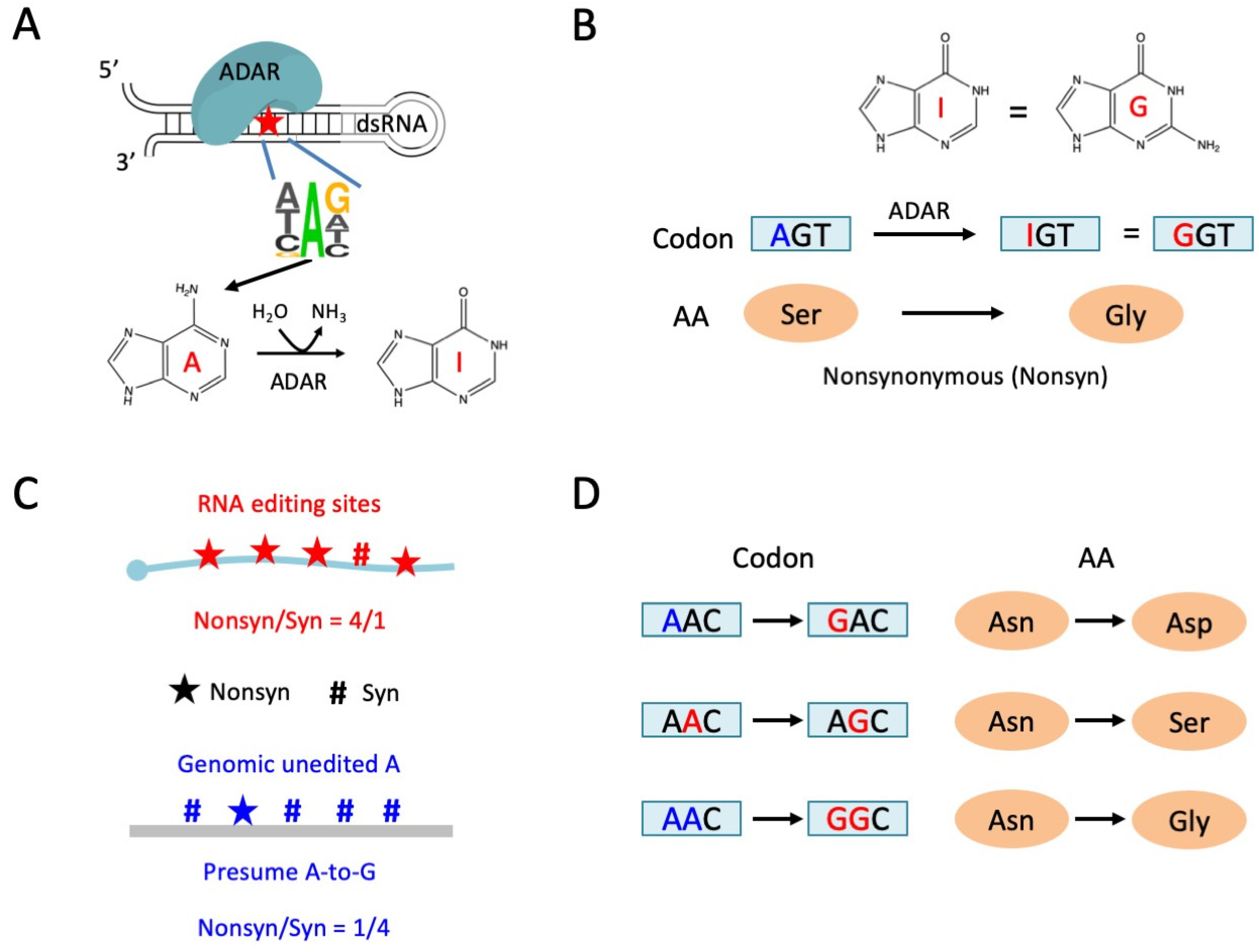
1.1. Adaptive A-to-I RNA Editing in Insects
1.2. Linkage of RNA Editing Events Provides Insights into the Functional Annotation of RNA Editing
1.3. Aims and Scopes
2. Materials and Methods
2.1. Data Collection
2.2. LD (Linkage Disequilibrium) Analysis
2.3. Statistics
3. Results
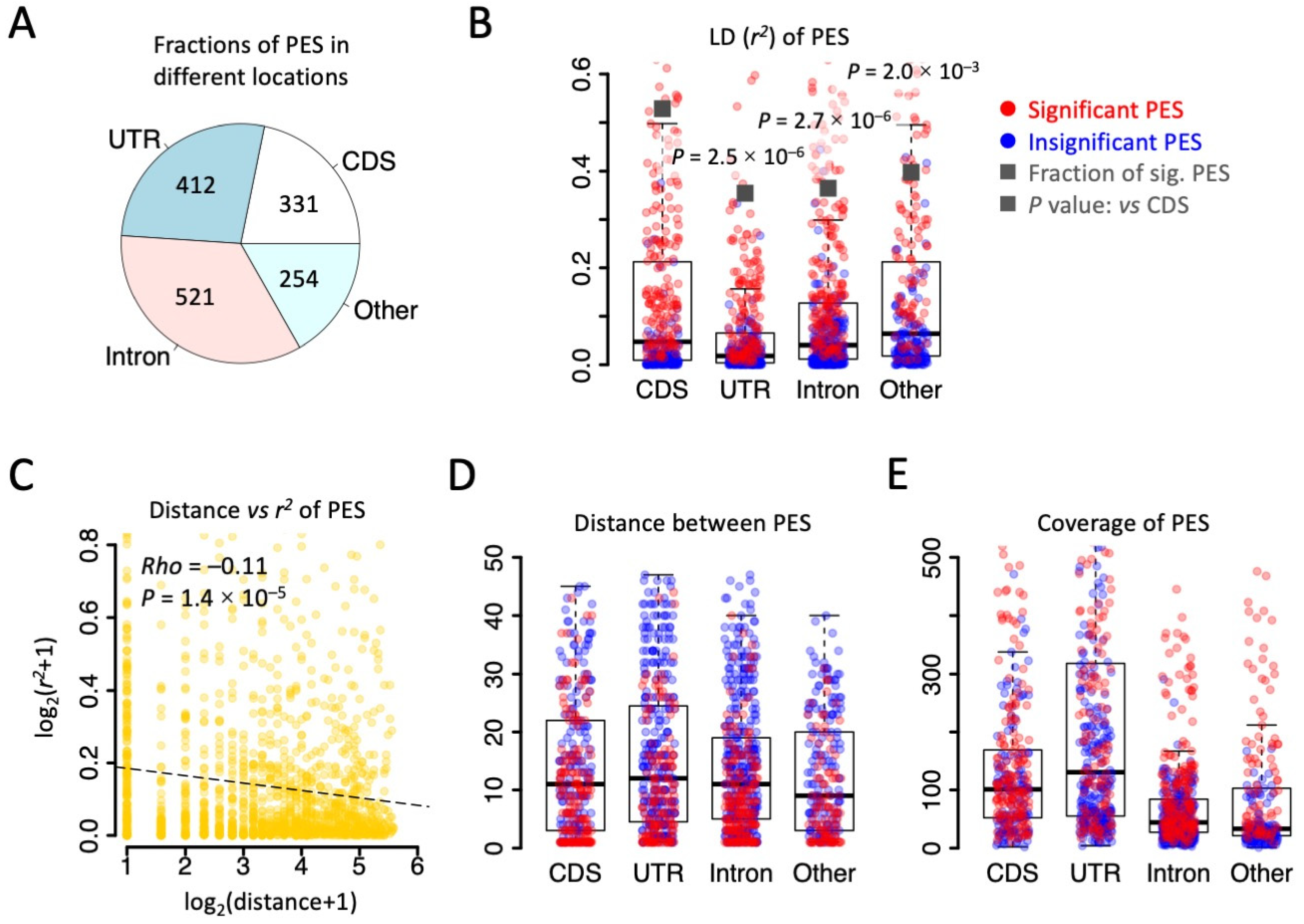
3.1. Wide-Spread Linkage of RNA Editing Events in the Drosophila Transcriptome
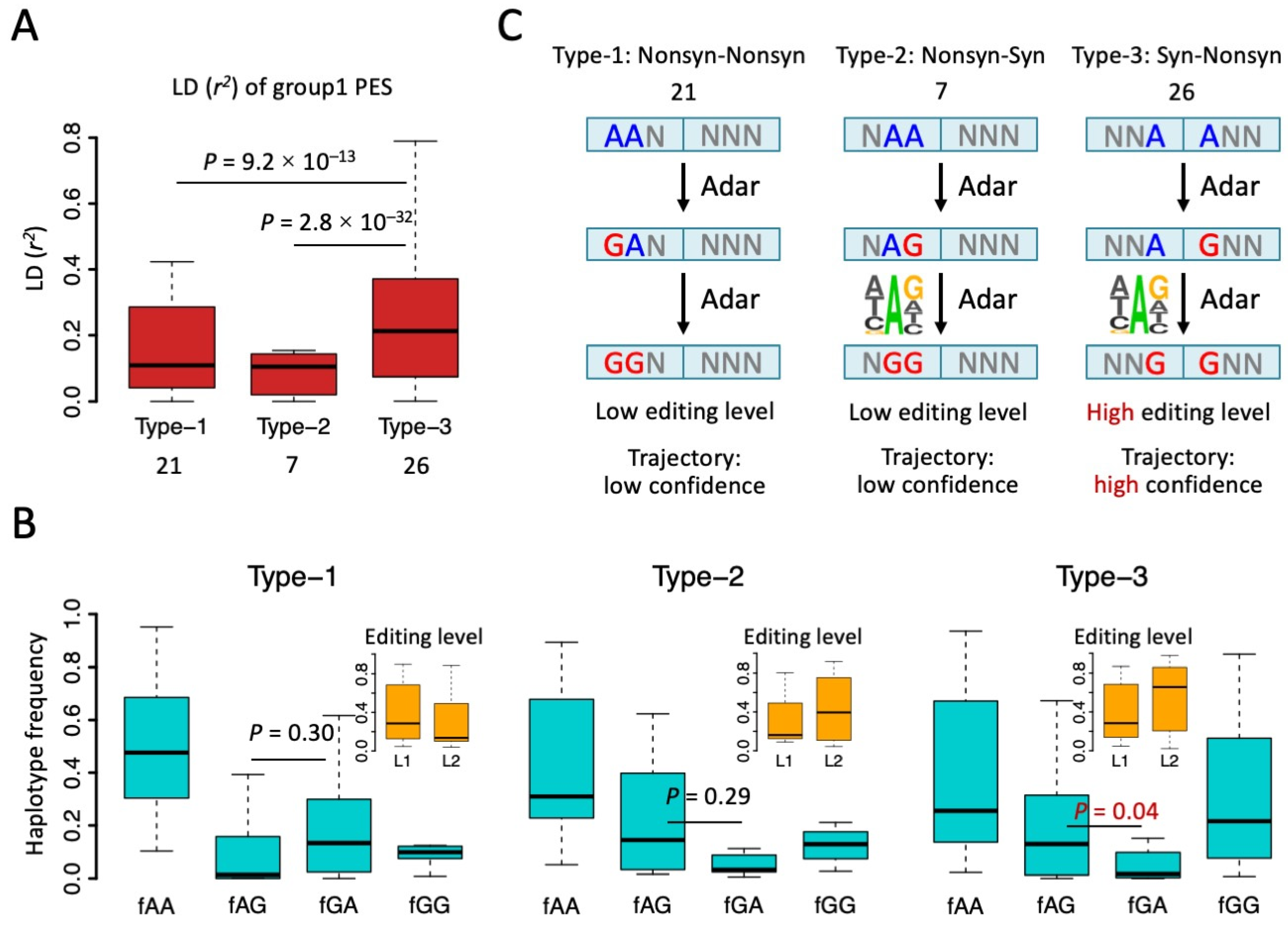
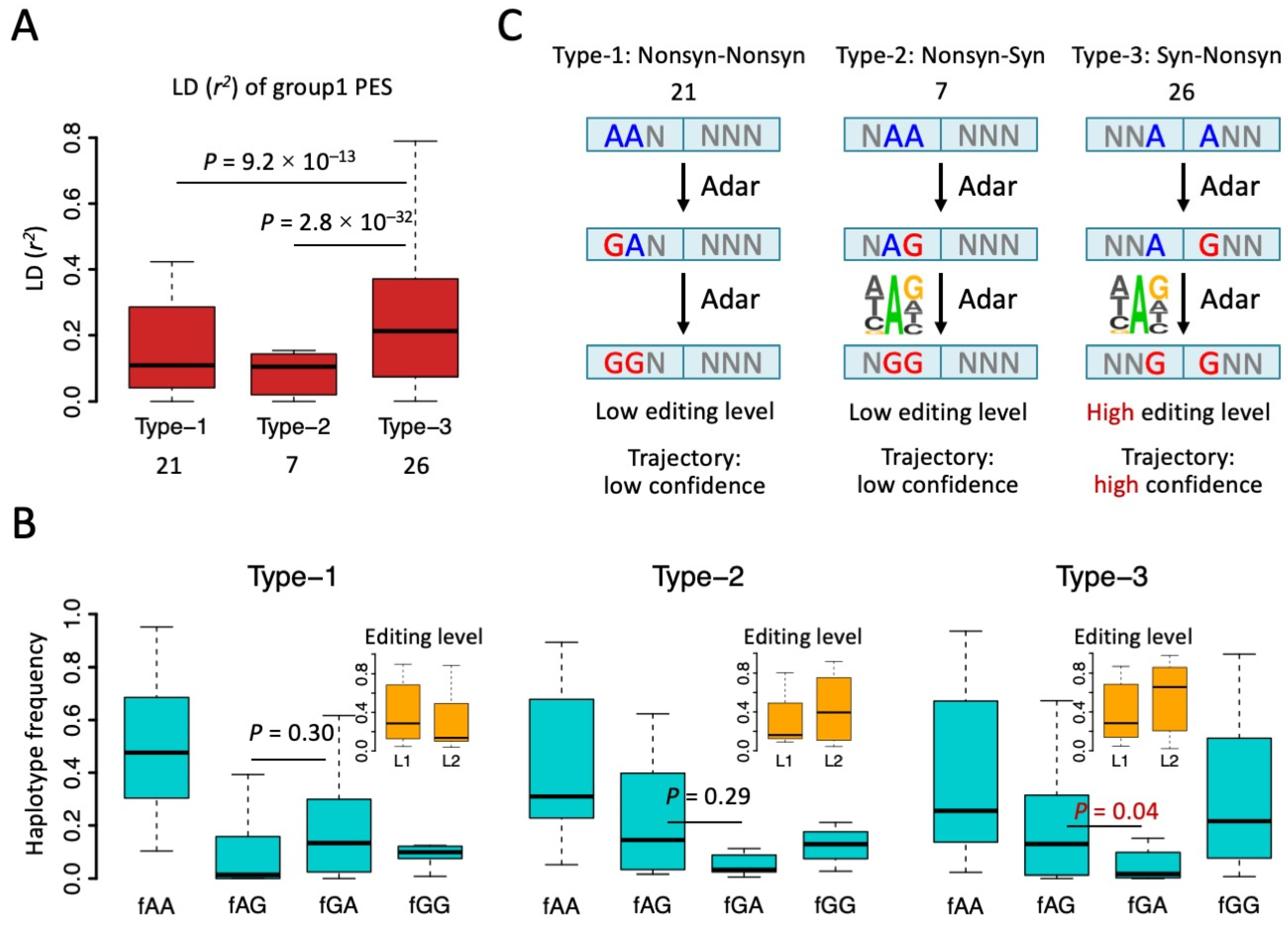
3.2. Biased Composition of Adjacent PESs Suggests Potential Interaction between Editing Sites
3.3. Haplotype Frequency Suggests Many Synonymous Editing Sites Are Passengers
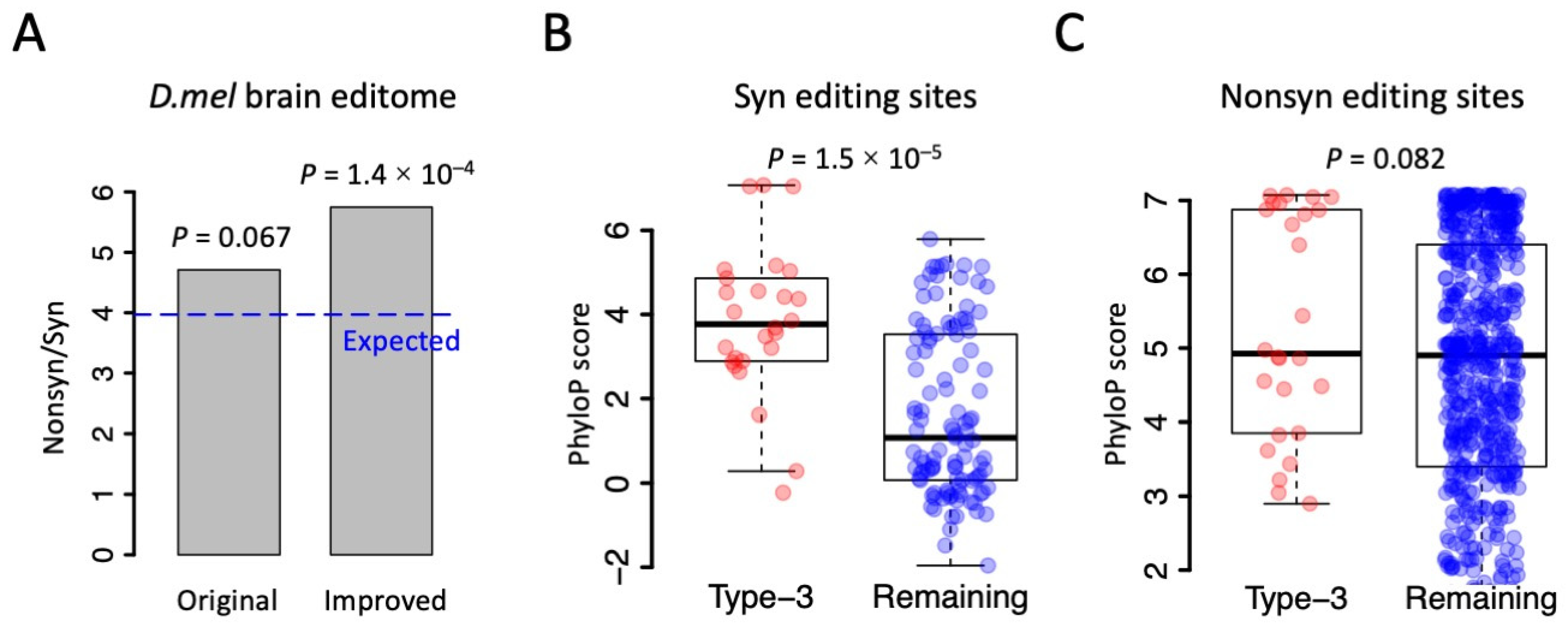
3.4. Underestimation of Adaptive Signals of RNA Editing Due to the Hitchhiking of Synonymous Sites
3.5. Synonymous Sites in Type-3 PESs Were More Conserved than Other Synonymous Editing Sites
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADAR | adenosine deaminase acting on RNA |
| A-to-I | adenosine-to-inosine |
| CDS | coding sequence |
| LD | linkage disequilibrium |
| Nonsyn | nonsynonymous |
| PES | pair of editing sites |
| Syn | synonymous |
| VCF | variant calling format |
References
- Zhang, P.; Zhu, Y.; Guo, Q.; Li, J.; Zhan, X.; Yu, H.; Xie, N.; Tan, H.; Lundholm, N.; Garcia-Cuetos, L.; et al. On the origin and evolution of RNA editing in metazoans. Cell Rep. 2023, 42, 112112. [Google Scholar] [CrossRef] [PubMed]
- Hung, L.Y.; Chen, Y.J.; Mai, T.L.; Chen, C.Y.; Yang, M.Y.; Chiang, T.W.; Wang, Y.D.; Chuang, T.J. An evolutionary landscape of A-to-I RNA editome across metazoan species. Genome Biol. Evol. 2018, 10, 521–537. [Google Scholar] [CrossRef] [PubMed]
- Porath, H.T.; Knisbacher, B.A.; Eisenberg, E.; Levanon, E.Y. Massive A-to-I RNA editing is common across the metazoa and correlates with dsRNA abundance. Genome Biol. 2017, 18, 185. [Google Scholar] [CrossRef] [PubMed]
- Savva, Y.A.; Rieder, L.E.; Reenan, R.A. The ADAR protein family. Genome Biol. 2012, 13, 252. [Google Scholar] [CrossRef]
- Bass, B.L.; Weintraub, H. An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell 1988, 55, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Ma, L.; Song, F.; Tian, L.; Cai, W.; Li, H. Autorecoding A-to-I RNA editing sites in the Adar gene underwent compensatory gains and losses in major insect clades. RNA 2023, 29, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, Z.; Lian, J.; Schiott, M.; Jin, L.; Zhang, P.; Zhang, Y.; Nygaard, S.; Peng, Z.; Zhou, Y.; et al. Caste-specific RNA editomes in the leaf-cutting ant Acromyrmex echinatior. Nat. Commun. 2014, 5, 4943. [Google Scholar] [CrossRef]
- Liscovitch-Brauer, N.; Alon, S.; Porath, H.T.; Elstein, B.; Unger, R.; Ziv, T.; Admon, A.; Levanon, E.Y.; Rosenthal, J.J.C.; Eisenberg, E. Trade-off between transcriptome plasticity and genome evolution in cephalopods. Cell 2017, 169, 191–202.e111. [Google Scholar] [CrossRef]
- Tan, M.H.; Li, Q.; Shanmugam, R.; Piskol, R.; Kohler, J.; Young, A.N.; Liu, K.I.; Zhang, R.; Ramaswami, G.; Ariyoshi, K.; et al. Dynamic landscape and regulation of RNA editing in mammals. Nature 2017, 550, 249–254. [Google Scholar] [CrossRef]
- Martin, F.H.; Castro, M.M.; Aboul-ela, F.; Tinoco, I., Jr. Base pairing involving deoxyinosine: Implications for probe design. Nucleic Acids Res. 1985, 13, 8927–8938. [Google Scholar] [CrossRef]
- Eisenberg, E.; Levanon, E.Y. A-to-I RNA editing—Immune protector and transcriptome diversifier. Nat. Rev. Genet. 2018, 19, 473–490. [Google Scholar] [CrossRef]
- Sommer, B.; Kohler, M.; Sprengel, R.; Seeburg, P.H. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell 1991, 67, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Egebjerg, J.; Heinemann, S.F. Ca2+ permeability of unedited and edited versions of the kainate selective glutamate receptor GluR6. Proc. Natl. Acad. Sci. USA 1993, 90, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, M.; Single, F.N.; Kohler, M.; Sommer, B.; Sprengel, R.; Seeburg, P.H. RNA editing of ampa receptor subunit Glur-B—A base-paired intron-exon structure determines position and efficiency. Cell 1993, 75, 1361–1370. [Google Scholar] [CrossRef]
- Tonkin, L.A.; Saccomanno, L.; Morse, D.P.; Brodigan, T.; Krause, M.; Bass, B.L. RNA editing by ADARs is important for normal behavior in Caenorhabditis elegans. EMBO J. 2002, 21, 6025–6035. [Google Scholar] [CrossRef] [PubMed]
- Palladino, M.J.; Keegan, L.P.; O’Connell, M.A.; Reenan, R.A. A-to-I pre-mRNA editing in Drosophila is primarily involved in adult nervous system function and integrity. Cell 2000, 102, 437–449. [Google Scholar] [CrossRef]
- Picardi, E.; Pesole, G. REDItools: High-throughput RNA editing detection made easy. Bioinformatics 2013, 29, 1813–1814. [Google Scholar] [CrossRef]
- Ramaswami, G.; Li, J.B. RADAR: A rigorously annotated database of A-to-I RNA editing. Nucleic Acids Res. 2014, 42, D109–D113. [Google Scholar] [CrossRef]
- Porath, H.T.; Hazan, E.; Shpigler, H.; Cohen, M.; Band, M.; Ben-Shahar, Y.; Levanon, E.Y.; Eisenberg, E.; Bloch, G. RNA editing is abundant and correlates with task performance in a social bumblebee. Nat. Commun. 2019, 10, 1605. [Google Scholar] [CrossRef]
- Duan, Y.; Xu, Y.; Song, F.; Tian, L.; Cai, W.; Li, H. Differential adaptive RNA editing signals between insects and plants revealed by a new measurement termed haplotype diversity. Biol. Direct 2023, 18, 47. [Google Scholar] [CrossRef]
- Gommans, W.M.; Mullen, S.P.; Maas, S. RNA editing: A driving force for adaptive evolution? Bioessays 2009, 31, 1137–1145. [Google Scholar] [CrossRef]
- Xin, K.; Zhang, Y.; Fan, L.; Qi, Z.; Feng, C.; Wang, Q.; Jiang, C.; Xu, J.R.; Liu, H. Experimental evidence for the functional importance and adaptive advantage of A-to-I RNA editing in fungi. Proc. Natl. Acad. Sci. USA 2023, 120, e2219029120. [Google Scholar] [CrossRef]
- Birk, M.A.; Liscovitch-Brauer, N.; Dominguez, M.J.; McNeme, S.; Yue, Y.; Hoff, J.D.; Twersky, I.; Verhey, K.J.; Sutton, R.B.; Eisenberg, E.; et al. Temperature-dependent RNA editing in octopus extensively recodes the neural proteome. Cell 2023, 186, 2544–2555 e2513. [Google Scholar] [CrossRef]
- Rangan, K.J.; Reck-Peterson, S.L. RNA recoding in cephalopods tailors microtubule motor protein function. Cell 2023, 186, 2531–2543.e2511. [Google Scholar] [CrossRef] [PubMed]
- Yablonovitch, A.L.; Fu, J.; Li, K.; Mahato, S.; Kang, L.; Rashkovetsky, E.; Korol, A.B.; Tang, H.; Michalak, P.; Zelhof, A.C.; et al. Regulation of gene expression and RNA editing in Drosophila adapting to divergent microclimates. Nat. Commun. 2017, 8, 1570. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zheng, C.; Xu, S.; Xu, Y.; Song, F.; Tian, L.; Cai, W.; Li, H.; Duan, Y. A full repertoire of Hemiptera genomes reveals a multi-step evolutionary trajectory of auto-RNA editing site in insect Adar gene. RNA Biol. 2023, 20, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Alon, S.; Garrett, S.C.; Levanon, E.Y.; Olson, S.; Graveley, B.R.; Rosenthal, J.J.; Eisenberg, E. The majority of transcripts in the squid nervous system are extensively recoded by A-to-I RNA editing. eLife 2015, 4, 198. [Google Scholar] [CrossRef] [PubMed]
- Sapiro, A.L.; Shmueli, A.; Henry, G.L.; Li, Q.; Shalit, T.; Yaron, O.; Paas, Y.; Li, J.B.; Shohat-Ophir, G. Illuminating spatial A-to-I RNA editing signatures within the Drosophila brain. Proc. Natl. Acad. Sci. USA 2019, 116, 2318–2327. [Google Scholar] [CrossRef]
- Licht, K.; Kapoor, U.; Amman, F.; Picardi, E.; Martin, D.; Bajad, P.; Jantsch, M.F. A high resolution A-to-I editing map in the mouse identifies editing events controlled by pre-mRNA splicing. Genome Res. 2019, 29, 1453–1463. [Google Scholar] [CrossRef]
- Graveley, B.R.; Brooks, A.N.; Carlson, J.W.; Duff, M.O.; Landolin, J.M.; Yang, L.; Artieri, C.G.; van Baren, M.J.; Boley, N.; Booth, B.W.; et al. The developmental transcriptome of Drosophila melanogaster. Nature 2011, 471, 473–479. [Google Scholar] [CrossRef]
- Yu, Y.; Zhou, H.; Kong, Y.; Pan, B.; Chen, L.; Wang, H.; Hao, P.; Li, X. The landscape of A-to-I RNA editome is shaped by both positive and purifying selection. PLoS Genet. 2016, 12, e1006191. [Google Scholar] [CrossRef]
- Yablonovitch, A.L.; Deng, P.; Jacobson, D.; Li, J.B. The evolution and adaptation of A-to-I RNA editing. PLoS Genet. 2017, 13, e1007064. [Google Scholar] [CrossRef]
- Zhang, R.; Deng, P.; Jacobson, D.; Li, J.B. Evolutionary analysis reveals regulatory and functional landscape of coding and non-coding RNA editing. PLoS Genet. 2017, 13, e1006563. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.; Menet, J.S.; Rosbash, M. Nascent-seq indicates widespread cotranscriptional RNA editing in Drosophila. Mol. Cell 2012, 47, 27–37. [Google Scholar] [CrossRef] [PubMed]
- St Laurent, G.; Tackett, M.R.; Nechkin, S.; Shtokalo, D.; Antonets, D.; Savva, Y.A.; Maloney, R.; Kapranov, P.; Lawrence, C.E.; Reenan, R.A. Genome-wide analysis of A-to-I RNA editing by single-molecule sequencing in Drosophila. Nat. Struct. Mol. Biol. 2013, 20, 1333–1339. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Li, H.; Cai, W. Adaptation of A-to-I RNA editing in bacteria, fungi, and animals. Front. Microbiol. 2023, 14, 1204080. [Google Scholar] [CrossRef]
- Porath, H.T.; Carmi, S.; Levanon, E.Y. A genome-wide map of hyper-edited RNA reveals numerous new sites. Nat. Commun. 2014, 5, 4726. [Google Scholar] [CrossRef]
- Lewontin, R.C. On measures of gametic disequilibrium. Genetics 1988, 120, 849–852. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Adams, M.D.; Celniker, S.E.; Holt, R.A.; Evans, C.A.; Gocayne, J.D.; Amanatides, P.G.; Scherer, S.E.; Li, P.W.; Hoskins, R.A.; Galle, R.F.; et al. The genome sequence of Drosophila melanogaster. Science 2000, 287, 2185–2195. [Google Scholar] [CrossRef]
- Duan, Y.; Cai, W.; Li, H. Chloroplast C-to-U RNA editing in vascular plants is adaptive due to its restorative effect: Testing the restorative hypothesis. RNA 2023, 29, 141–152. [Google Scholar] [CrossRef]
- Xu, G.; Zhang, J. Human coding RNA editing is generally nonadaptive. Proc. Natl. Acad. Sci. USA 2014, 111, 3769–3774. [Google Scholar] [CrossRef]
- Jiang, D.; Zhang, J. The preponderance of nonsynonymous A-to-I RNA editing in coleoids is nonadaptive. Nat. Commun. 2019, 10, 5411. [Google Scholar] [CrossRef]
- Shoshan, Y.; Liscovitch-Brauer, N.; Rosenthal, J.J.C.; Eisenberg, E. Adaptive proteome diversification by nonsynonymous A-to-I RNA editing in coleoid cephalopods. Mol. Biol. Evol. 2021, 38, 3775–3788. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Zhang, J. In search of beneficial coding RNA editing. Mol. Biol. Evol. 2015, 32, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Quinones-Valdez, G.; Fu, T.; Huang, E.; Choudhury, M.; Reese, F.; Mortazavi, A.; Xiao, X. L-GIREMI uncovers RNA editing sites in long-read RNA-seq. Genome Biol. 2023, 24, 171. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Duan, Y. Genome-Wide Analysis on Driver and Passenger RNA Editing Sites Suggests an Underestimation of Adaptive Signals in Insects. Genes 2023, 14, 1951. https://doi.org/10.3390/genes14101951
Zhang Y, Duan Y. Genome-Wide Analysis on Driver and Passenger RNA Editing Sites Suggests an Underestimation of Adaptive Signals in Insects. Genes. 2023; 14(10):1951. https://doi.org/10.3390/genes14101951
Chicago/Turabian StyleZhang, Yuchen, and Yuange Duan. 2023. "Genome-Wide Analysis on Driver and Passenger RNA Editing Sites Suggests an Underestimation of Adaptive Signals in Insects" Genes 14, no. 10: 1951. https://doi.org/10.3390/genes14101951
APA StyleZhang, Y., & Duan, Y. (2023). Genome-Wide Analysis on Driver and Passenger RNA Editing Sites Suggests an Underestimation of Adaptive Signals in Insects. Genes, 14(10), 1951. https://doi.org/10.3390/genes14101951