
A Sporadic Case of COL1A1 Osteogenesis Imperfecta: From Prenatal Diagnosis to Outcomes in Infancy—Case Report and Literature Review

 , ,
, ,
Abstract
:1. Introduction
2. Case Report
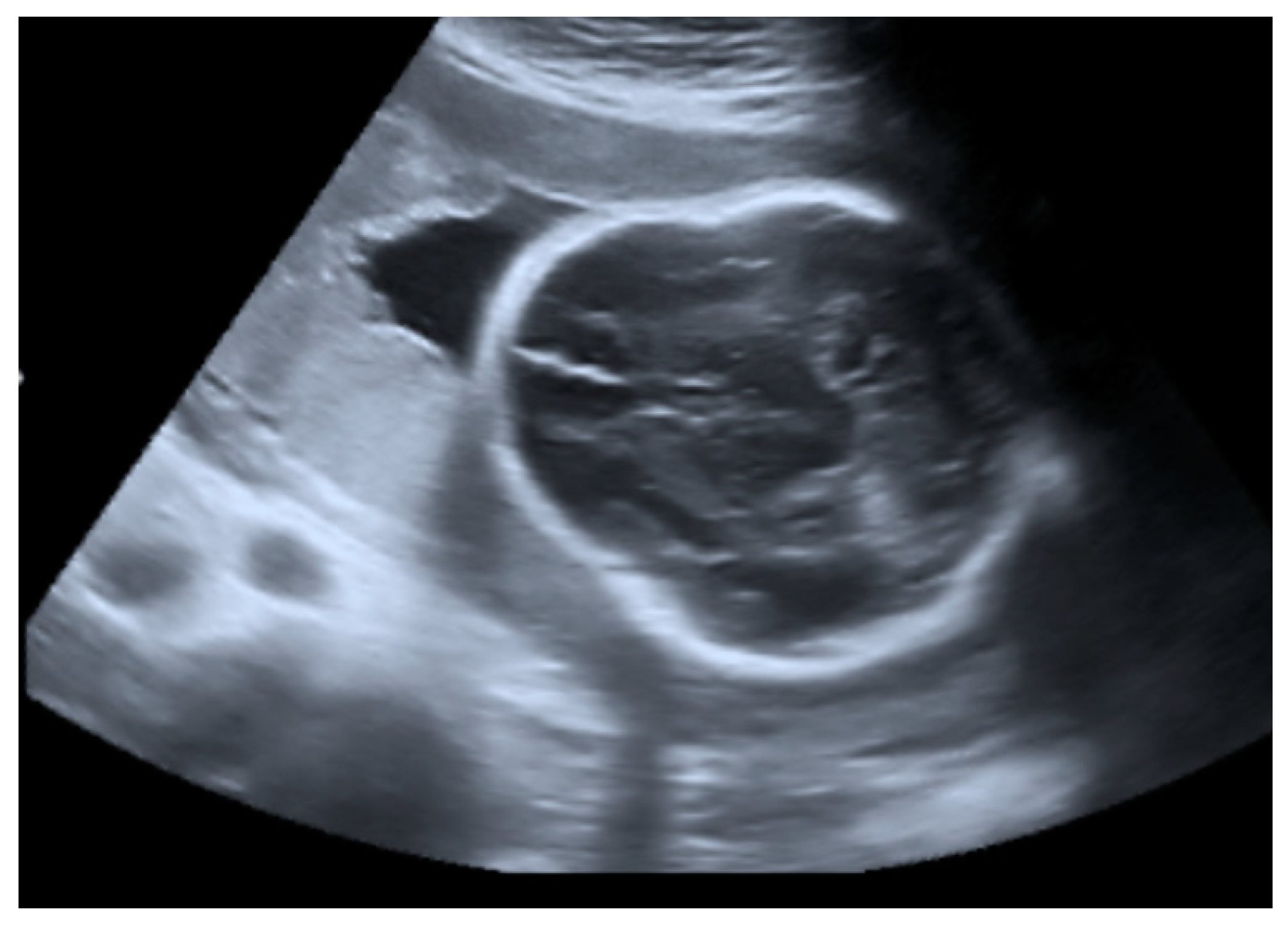
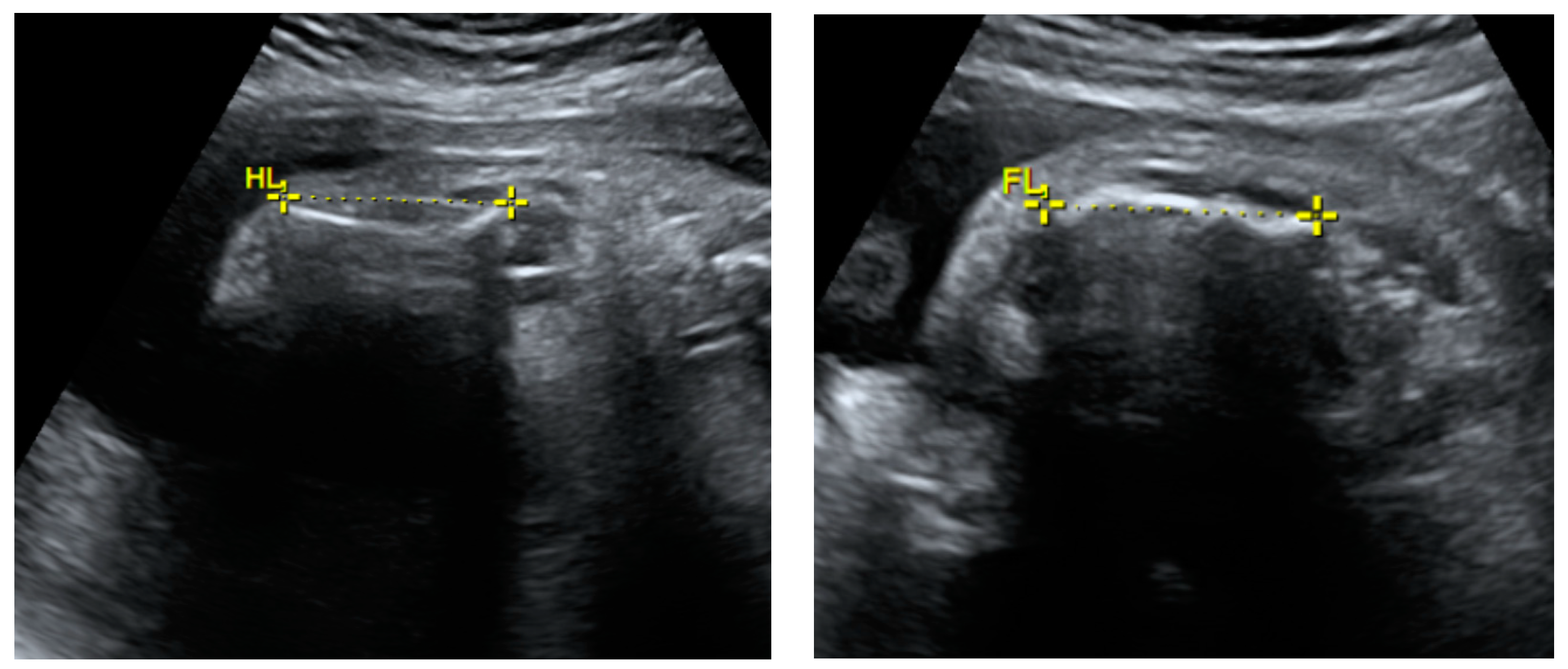
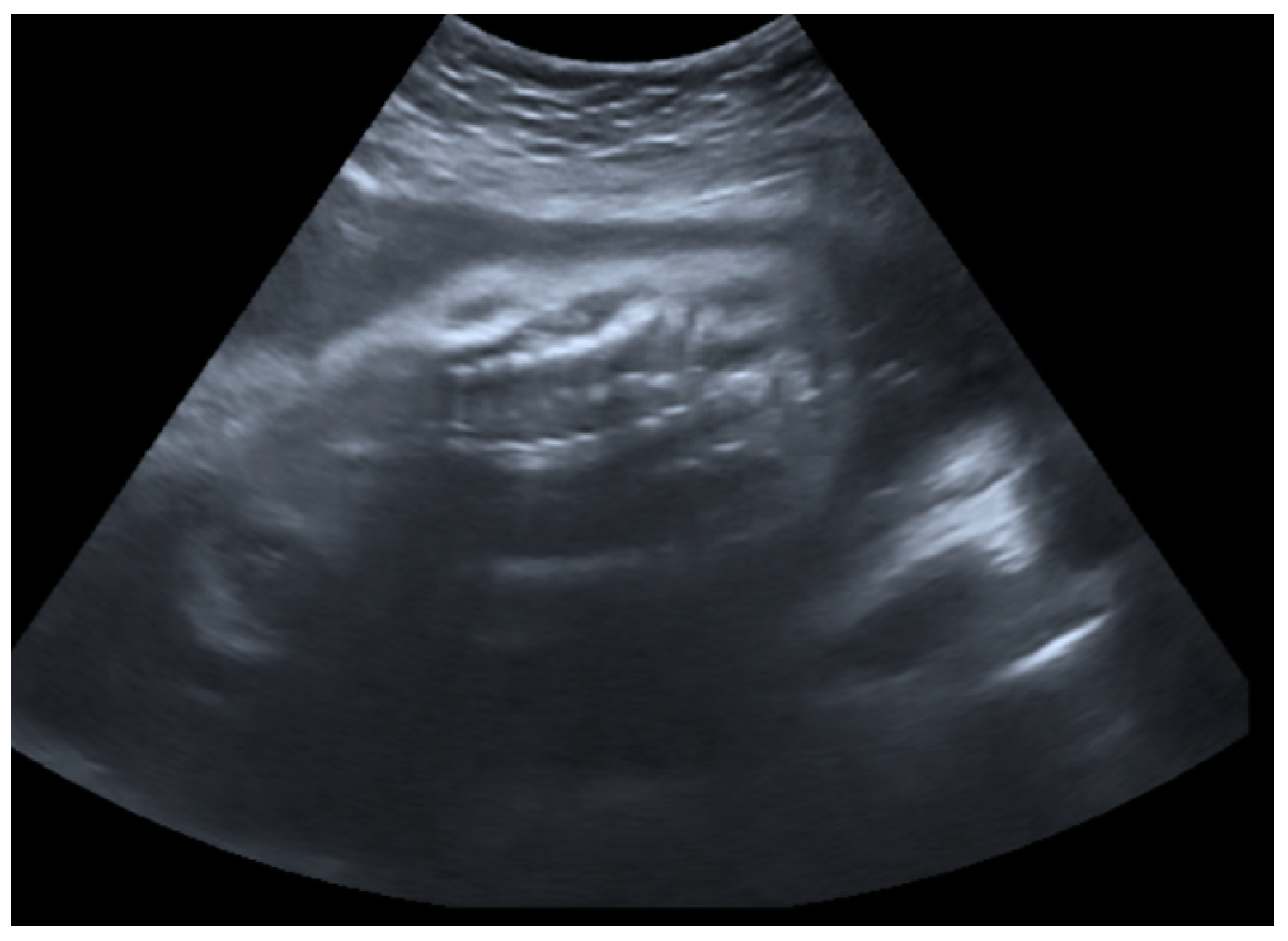
2.1. Prenatal Findings and Further Pregnancy Care
2.2. Birth and Early Treatment
2.3. Further Patient Investigations, Management, and Treatment after Birth
2.4. Follow-Up and Outcome One Year after Birth
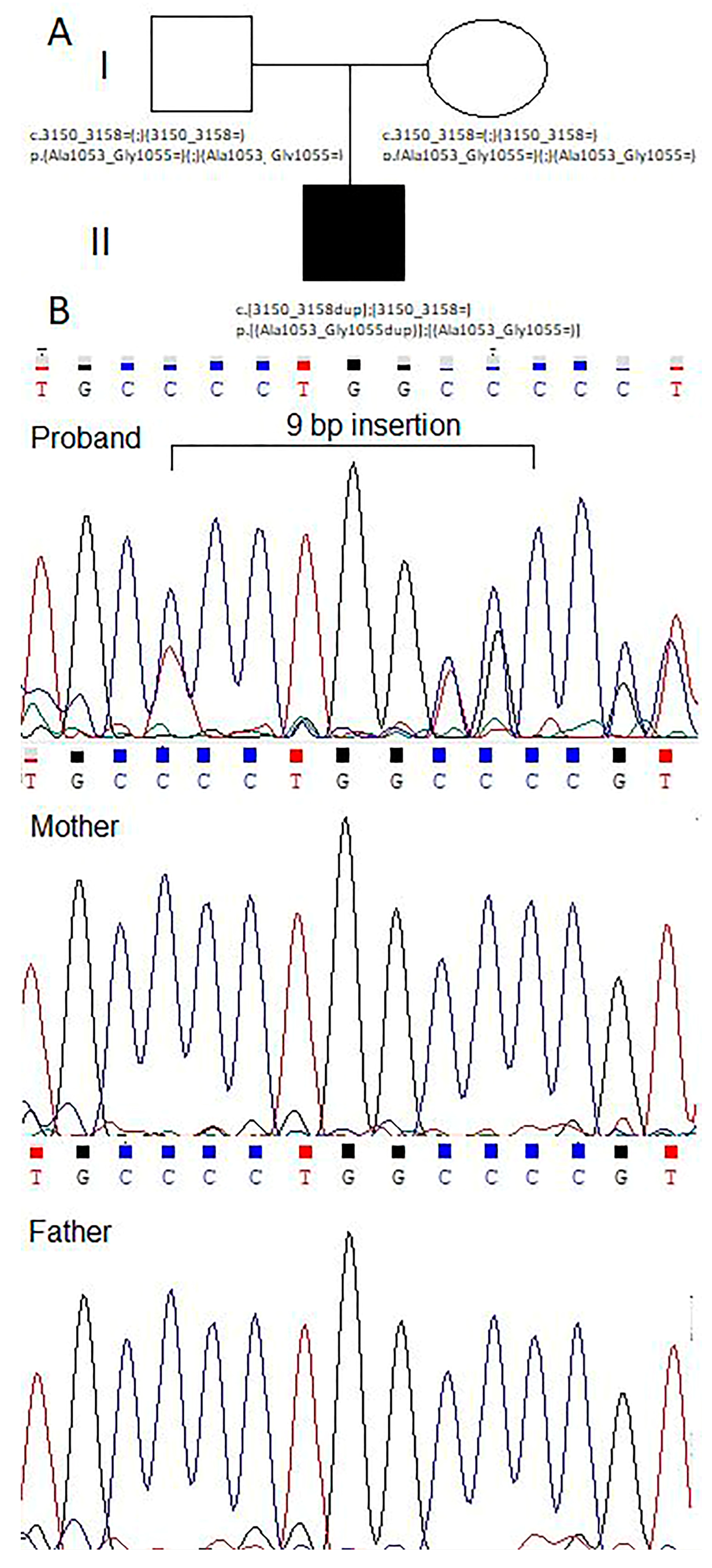
2.5. Genetic Counseling
3. Discussion and Literature Review
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marini, J. Nelson Textbook of Pediatrics; Behrman, R.E., Kliegman, R.M., Jensen, R.M., Eds.; Saunders: Philadelphia, PA, USA, 2004. [Google Scholar]
- Marini, J.C.; Reich, A.; Smith, S.M. Osteogenesis imperfecta due to mutations in non-collagenous genes: Lessons in the biology of bone formation. Curr. Opin. Pediatr. 2014, 26, 500–507. [Google Scholar] [CrossRef]
- Marini, J.C.; Dang, A.N. Osteogenesis Imperfecta. Endotext. Updated 26 July 2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK279109/ (accessed on 2 September 2023).
- Rare Disease Database. Osteogenesis Imperfecta. Available online: https://rarediseases.org/rare-diseases/osteogenesis-imperfecta/ (accessed on 2 September 2023).
- Online Mendelian Inheritance in Man (OMIM). Osteogenesis Imperfecta. Available online: https://www.omim.org/search?index=entry&start=1&limit=10&sort=score+desc%2C+prefix_sort+desc&search=osteogenesis+imperfecta (accessed on 2 September 2023).
- Available online: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=666 (accessed on 2 September 2023).
- Steiner, R.D.; Basel, D. COL1A1/2 Osteogenesis Imperfecta; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2005. [Google Scholar]
- Sam, J.E.; Dharmalingam, M. Osteogenesis Imperfecta. Indian. J. Endocrinol. Metab. 2017, 21, 903–908. [Google Scholar] [CrossRef]
- Parilla, B.V.; Leeth, E.A.; Kambich, M.P.; Chilis, P.; MacGregor, S.N. Antenatal Detection of Skeletal Dysplasias. J. Ultrasound Med. 2003, 22, 255–258. [Google Scholar] [CrossRef]
- Pace, J.M.; Atkinson, M.; Willing, M.C.; Wallis, G.; Byers, P.H. Deletions and duplications of Gly-Xaa-Yaa triplet repeats in the triple helical domains of type I collagen chains disrupt helix formation and result in several types of osteogenesis imperfecta. Hum. Mutat. 2001, 18, 319–326. [Google Scholar] [CrossRef]
- Khalil, A.; Pajkrt, E.; Chitty, L.S. Early prenatal diagnosis of skeletal anomalies. Prenat. Diagn. 2011, 31, 115–124. [Google Scholar] [CrossRef]
- Krakow, D. Skeletal dysplasias. Clin. Perinatol. 2015, 42, 301–319. [Google Scholar] [CrossRef]
- Krakow, D.; Lachman, R.S.; Rimoin, D.L. Guidelines for the prenatal diagnosis of fetal skeletal dysplasias. Genet. Med. 2009, 11, 127–133. [Google Scholar] [CrossRef]
- Dey, S.; Roy, P. First Trimester Ultrasound Detection of Osteogenesis Imperfecta: Prenatal ultrasound clues discussed with a case report. J. Emerg. Technol. Innov. Res. 2023, 10, 527–532. [Google Scholar]
- Noel, A.-E.; Brown, R.N. Advances in evaluating the fetal skeleton. Int. J. Womens Health 2014, 6, 489–500. [Google Scholar] [CrossRef]
- French, T.; Savarirayan, R. Thanatophoric Dysplasia. In GeneReviews® [Internet]; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1366/ (accessed on 2 November 2023).
- Chang, B.; Keating, S.; Mikhael, M.; Lim, J. Osteogenesis Imperfecta: Multidisciplinary and Goal-Centered Care. AJP Rep. 2022, 12, e144–e147. [Google Scholar] [CrossRef]
- Luis, T.; Gonçalves, A.C.; Rodrigues, E.; Mendes, M.; Teixeira, T. Brittle Bone Disease: A Case Report. Cureus 2022, 14, e31259. [Google Scholar] [CrossRef]
- Canda, M.T.; Ceylaner, S.; Doganay Caglayan, L.; Demir, A.B.; Demir, N. Prenatal Diagnosis of Osteogenesis Imperfecta Type III. J. Obstet. Gynaecol. India 2019, 69, 374–376. [Google Scholar] [CrossRef]
- Bayram, S.; Mert, L.; Anarat, F.B.; Chodza, M.; Ergin, Ö.N. A Newborn with Multiple Fractures in Osteogenesis Imperfecta: A Case Report. J. Orthop. Case Rep. 2018, 8, 71–73. [Google Scholar] [CrossRef]
- Mbono Betoko, R.C.; Ngo Um Sap, S.; Ngo Yamben, M.-A.; Tony Nengom, J.; Koki Ndombo, P. Osteogenesis Imperfecta in neonatal period in Cameroon: A case report. Clin. Case Rep. 2020, 9, 526–530. [Google Scholar] [CrossRef]
- Wu, D.; Wang, Y.; Huang, H. A novel variant of the IFITM5 gene within the 5’-UTR causes neonatal transverse clavicular fracture: Expanding the genetic spectrum. Mol. Genet. Genom. Med. 2020, 8, e1287. [Google Scholar] [CrossRef]
- Rodriguez Celin, M.; Moosa, S.; Fano, V. Uncommon IFITM5 mutation associated with severe skeletal deformity in osteogenesis imperfecta. Ann. Hum. Genet. 2018, 82, 477–481. [Google Scholar] [CrossRef]
- Alhousseini, A.; Mahaseth, M.; Zeineddine, S.; Jaiman, S.; Berman, S.; Bryant, D.; Tan, S.; Hernandez-Andrade, E. A Non-Lethal Osteogenesis Imperfecta Type II Mutation. Gynecol. Obstet. Investig. 2018, 84, 204–208. [Google Scholar] [CrossRef]
- Deguchi, M.; Tsuji, S.; Katsura, D.; Kasahara, K.; Kimura, F.; Murakami, T. Current Overview of Osteogenesis Imperfecta. Medicina 2021, 57, 464. [Google Scholar] [CrossRef]
- Byers, P.H.; Krakow, D.; Nunes, M.E.; Pepin, M.; American college of medical genetics. Genetic evaluation of suspected osteogenesis imperfecta (OI). Genet. Med. 2006, 8, 383–388. [Google Scholar] [CrossRef]
- Bellur, S.; Jain, M.; Cuthbertson, D.; Krakow, D.; Shapiro, J.R.; Steiner, R.D.; Smith, P.A.; Bober, M.B.; Hart, T.; Krischer, J.; et al. Cesarean delivery is not associated with decreased at-birth fracture rates in osteogenesis imperfecta. Genet. Med. 2015, 18, 570–576. [Google Scholar] [CrossRef]
- Marr, C.; Seasman, A.; Bishop, N. Managing the patient with osteogenesis imperfecta: A multidisciplinary approach. J. Multidiscip. Healthc. 2017, 10, 145–155. [Google Scholar] [CrossRef]
- Storoni, S.; Treurniet, S.; Micha, D.; Celli, M.; Bugiani, M.; van den Aardweg, J.G.; Eekhoff, E.M.W. Pathophysiology of respiratory failure in patients with osteogenesis imperfecta: A systematic review. Ann. Med. 2021, 53, 1676–1687. [Google Scholar] [CrossRef]
- Folkestad, L. Mortality and morbidity in patients with osteogenesis imperfecta in Denmark. Dan. Med. J. 2018, 65, B5454. [Google Scholar]
- Chien, A.L.; Mu, E.W.; Kang, S. Chapter 30—Skin in Osteogenesis Imperfecta. In Osteogenesis Imperfecta; Shapiro, J.R., Byers, P.H., Glorieux, F.H., Sponseller, P.D., Eds.; Academic Press: San Diego, CA, USA, 2014; pp. 283–288. Available online: https://www.sciencedirect.com/science/article/pii/B9780123971654000307 (accessed on 2 September 2023).
- Botor, M.; Fus-Kujawa, A.; Uroczynska, M.; Stepien, K.L.; Galicka, A.; Gawron, K.; Sieron, A.L. Osteogenesis Imperfecta: Current and Prospective Therapies. Biomolecules 2021, 11, 1493. [Google Scholar] [CrossRef]
- Plotkin, H.; Rauch, F.; Bishop, N.J.; Montpetit, K.; Ruck-Gibis, J.; Travers, R.; Glorieux, F.H. Pamidronate Treatment of Severe Osteogenesis Imperfecta in Children under 3 Years of Age*. J. Clin. Endocrinol. Metab. 2000, 85, 1846–1850. [Google Scholar] [CrossRef]
- Narayanan, P. Denosumab: A comprehensive review. South Asian J. Cancer 2013, 2, 272–277. [Google Scholar] [CrossRef]
- Riminucci, M.; Remoli, C.; Robey, P.G.; Bianco, P. Stem cells and bone diseases: New tools, new perspective. Bone 2014, 70, 55–61. [Google Scholar] [CrossRef]
- Deyle, D.R.; Khan, I.F.; Ren, G.; Wang, P.-R.; Kho, J.; Schwarze, U.; Russell, D.W. Normal collagen and bone production by gene-targeted human osteogenesis imperfecta iPSCs. Mol. Ther. 2011, 20, 204–213. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Body Part | Description |
|---|---|
| Whole body | Disproportionate |
| Skull | Uneven contour; mobile skull vault; fragmented skull bones; wide spaces between cranial sutures; wide anterior fontanel |
| Ears | Very low-set |
| Neck | Short |
| Intermammillary distance | Wide |
| Umbilicus | Low-set |
| Long bones | Very short humerus and femurs |
| Hands | Swollen |
| Feet | Clubfoot |
| Year | Author | Familial Case | Ultrasound Findings | Phenotype Features after Birth | OI Type and Variant | Problems after Birth | Outcome |
|---|---|---|---|---|---|---|---|
| 2022 | Chang et al. [17] | No | US findings at 35 weeks of GA: Asymmetric IUGR, short and angulated long bones, breech position. | Dysmorphisms with brachycephaly; widely separated suture in the posterior aspect of the skull; triangular-shaped face; prominent forehead; blue and gray sclera; malar flattening; pointed chin; shortened and bowed extremities. | OI type III. Pathogenic variant in the COL1A2 gene. | Respiratory distress; nutrition problems; prolonged apneic events associated with bradycardia and desaturations; pain. | Died from respiratory failure at day 30 of life. |
| 2022 | Luis et al. [18] | No | Not suspected before birth. Normal US findings. | Bilateral clavicle fractures with left brachial paresis. After one week: edema in the right hip joint; asymmetry in the folds; bilateral diaphyseal fracture of the femur. | OI type XV. Likely pathogenic variant c.324dup (p.(Gly109Argfs*46)) in probably homozygous state in the WNT1 gene. | Bone fractures requiring hospitalization; pain. | – |
| 2019 | Canda et al. [19] | No | US findings at 17 weeks of GA: short, angulated, and bowed both femurs (< 5th percentile); shortened other limb bones. | – | OI type III. Pathogenic variant c.1588G>A (p.(Gly530Ser)) in the heterozygous state in the COL1A1 gene. | – | Pregnancy termination at 23 weeks of gestation due to unfavorable prognosis. |
| 2018 | Bayram et al. [20] | No | Not suspected before birth. Normal US findings. | Not suspected after birth. | OI was confirmed with genetic testing. Detailed information about genetic testing and OI type was not provided. | Two weeks after birth: bilateral clavicle, bilateral femur, left humerus fractures. | No new fractures at the follow-up at 8 months old. |
| 2021 | Betoko et al. [21] | Case 1: No | Case 1: Not suspected before birth. Normal US findings. | Case 1: Not suspected after birth. | Case 1: OI type III was suspected according to clinical findings. Genetic testing not yet available in the country. | Case 1: At 26 days of age: reduced mobility; frontal bossing; moderate respiratory distress, bowed legs, shortened limbs. Multiple diaphyseal fractures of long bones; bones were curved and demineralized. | Case 1: At 9 months old: growth restriction; tiered vertebral collapse from T12 to L3 with no spinal deformities. Treatment with bisphosphonates prescribed. |
| 2021 | Betoko et al. [21] | Case 2: No | Case 2: US findings at the third pregnancy trimester: bowing and shortening of both femurs. | Case 2: Deformed limbs. | Case 2: OI type II or III was suspected according to antenatal and clinical findings. Genetic testing not yet available in the country. | Case 2: Neonatal sepsis at the age of 2 days. At 37 days of age: reduced mobility; frontal bossing; blue sclerae; bowed legs; shortened limbs. Multiple diaphyseal fractures of the long bones; fracture of the sternum; bones are curved. | Case 2: At the age of 3 months: pain improved; bone callus formed. Oral vitamin D supplements prescribed. |
| 2020 | Wu et al. [22] | No | Not suspected before birth. Normal US findings. | Not suspected after birth. | OI type V. Pathogenic variant c.-9C>A in heterozygous state in the IFITM5 gene. | 2 days after birth: pain and irritability. A transverse right mid-clavicular fracture was observed in a chest X-ray. | No new fractures and healed clavicle fracture at the age of 1 month old. |
| 2018 | Celin et al. [23] | No | US findings at 30 weeks of GA: severe polyhydramnios; delayed skull ossification; shortening and bowing of the long bones. | Blue sclera; hypoplastic thorax; bowing of the limbs; multiple fractures that occurred in utero (fractures of ribs, clavicles, humerus, upper limbs, and multiple compression fractures of spine). | OI type V. Pathogenic variant c.119C>T (p.(Ser40Leu)) in the heterozygous state in the IFITM5 gene. | Multiple bone fractures; transient tachypnea of the newborn; pain; nutritional problems. | At the age of two: development and motor delay; blue sclera; dentinogenesis imperfecta; short stature; bowed and short limbs; deformed thorax; relative macrocephaly with wide anterior fontanel. At age of 5: scoliosis, worsening of thoracic and skull deformations. |
| 2019 | Alhousseini et al. [24] | No | US findings at 21 weeks of GA: shortened and bowed femurs; both humerus with old fractures and callus; both tibia and fibula bowed. | Neonatal X-ray revealed: new and old healed fractures of extremities including the femur bones and humerus. Undermineralized extremities. | OI type II. Pathogenic variant c.1840G>C (p.(Gly614Arg)) in heterozygous state in the COL1A1 gene. | Respiratory distress requiring oxygen therapy; pain. | At the age of 22 months: mild bilateral conductive hearing loss; no new fractures or seizures since birth; dentinogenesis imperfecta; short and bowed limbs; able to sit on her own; not able to walk. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vankevičienė, K.; Matulevičienė, A.; Mazgelytė, E.; Paliulytė, V.; Vankevičienė, R.; Ramašauskaitė, D. A Sporadic Case of COL1A1 Osteogenesis Imperfecta: From Prenatal Diagnosis to Outcomes in Infancy—Case Report and Literature Review. Genes 2023, 14, 2062. https://doi.org/10.3390/genes14112062
Vankevičienė K, Matulevičienė A, Mazgelytė E, Paliulytė V, Vankevičienė R, Ramašauskaitė D. A Sporadic Case of COL1A1 Osteogenesis Imperfecta: From Prenatal Diagnosis to Outcomes in Infancy—Case Report and Literature Review. Genes. 2023; 14(11):2062. https://doi.org/10.3390/genes14112062
Chicago/Turabian StyleVankevičienė, Karolina, Aušra Matulevičienė, Eglė Mazgelytė, Virginija Paliulytė, Ramunė Vankevičienė, and Diana Ramašauskaitė. 2023. "A Sporadic Case of COL1A1 Osteogenesis Imperfecta: From Prenatal Diagnosis to Outcomes in Infancy—Case Report and Literature Review" Genes 14, no. 11: 2062. https://doi.org/10.3390/genes14112062
APA StyleVankevičienė, K., Matulevičienė, A., Mazgelytė, E., Paliulytė, V., Vankevičienė, R., & Ramašauskaitė, D. (2023). A Sporadic Case of COL1A1 Osteogenesis Imperfecta: From Prenatal Diagnosis to Outcomes in Infancy—Case Report and Literature Review. Genes, 14(11), 2062. https://doi.org/10.3390/genes14112062