
Genome Assembly and Microsatellite Marker Development Using Illumina and PacBio Sequencing in the Carex pumila (Cyperaceae) from Korea


Abstract
:1. Introduction
2. Materials and Methods
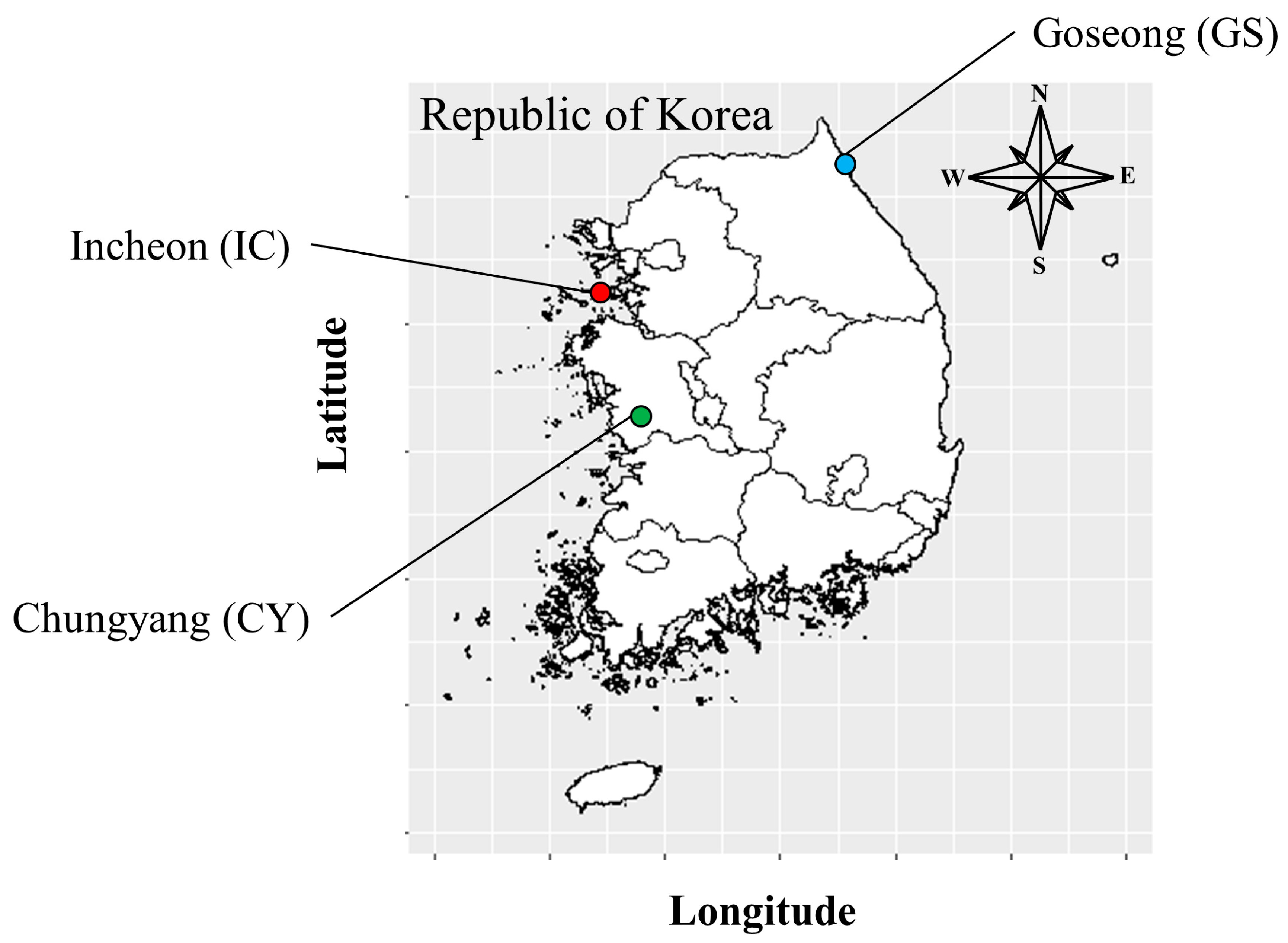
2.1. Sample Preparation
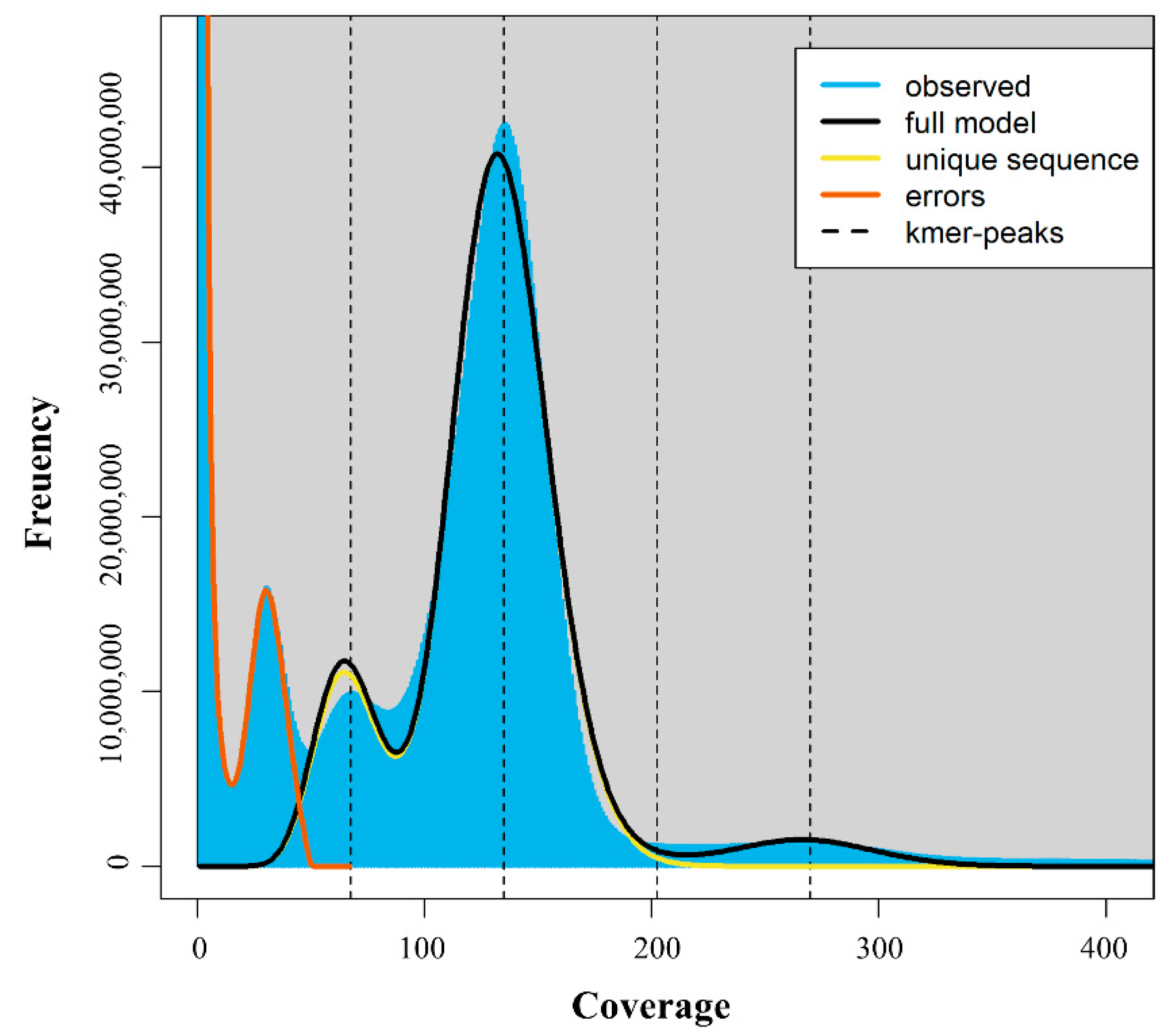
2.2. Genome Sequencing and De Novo Assembly
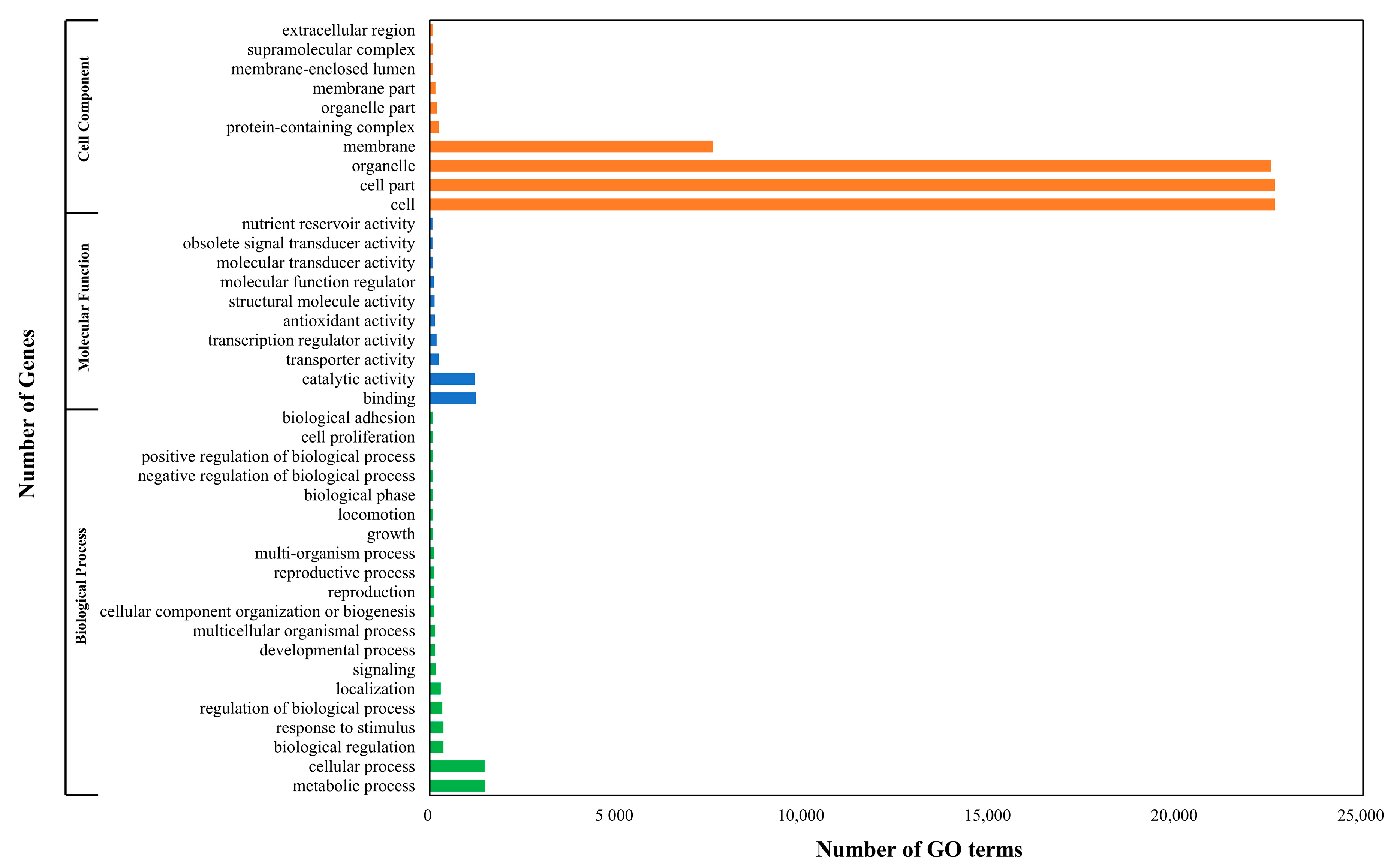
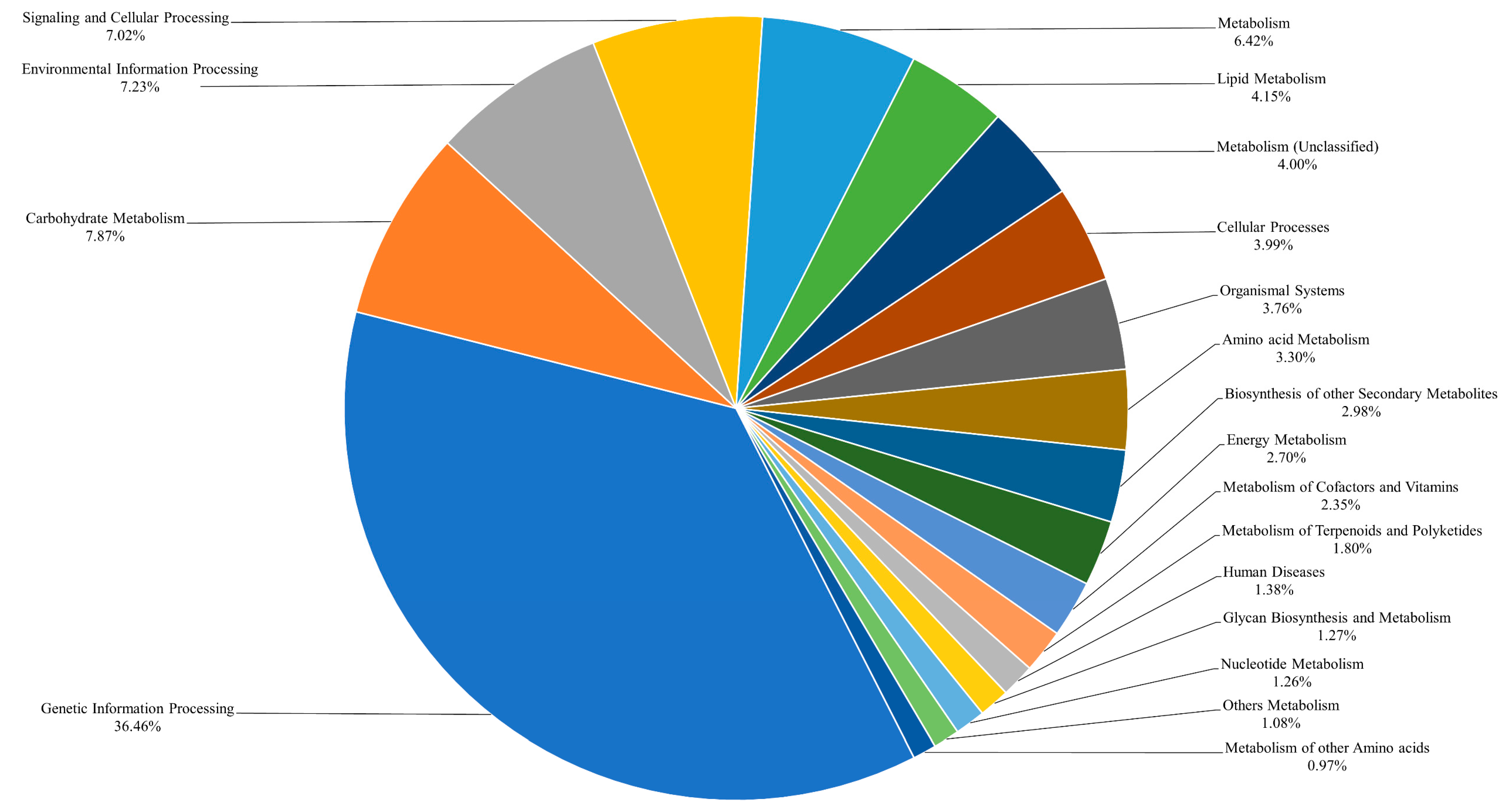
2.3. Genome Annotations and Gene Ontology Analysis
2.4. Phylogenetic Tree Reconstruction
2.5. Analysis of Microsatellite Markers and Genotyping
3. Results and Discussion
3.1. Genome Assembly of C. pumila
3.2. Annotation of Candidate Genes and Protein Prediction
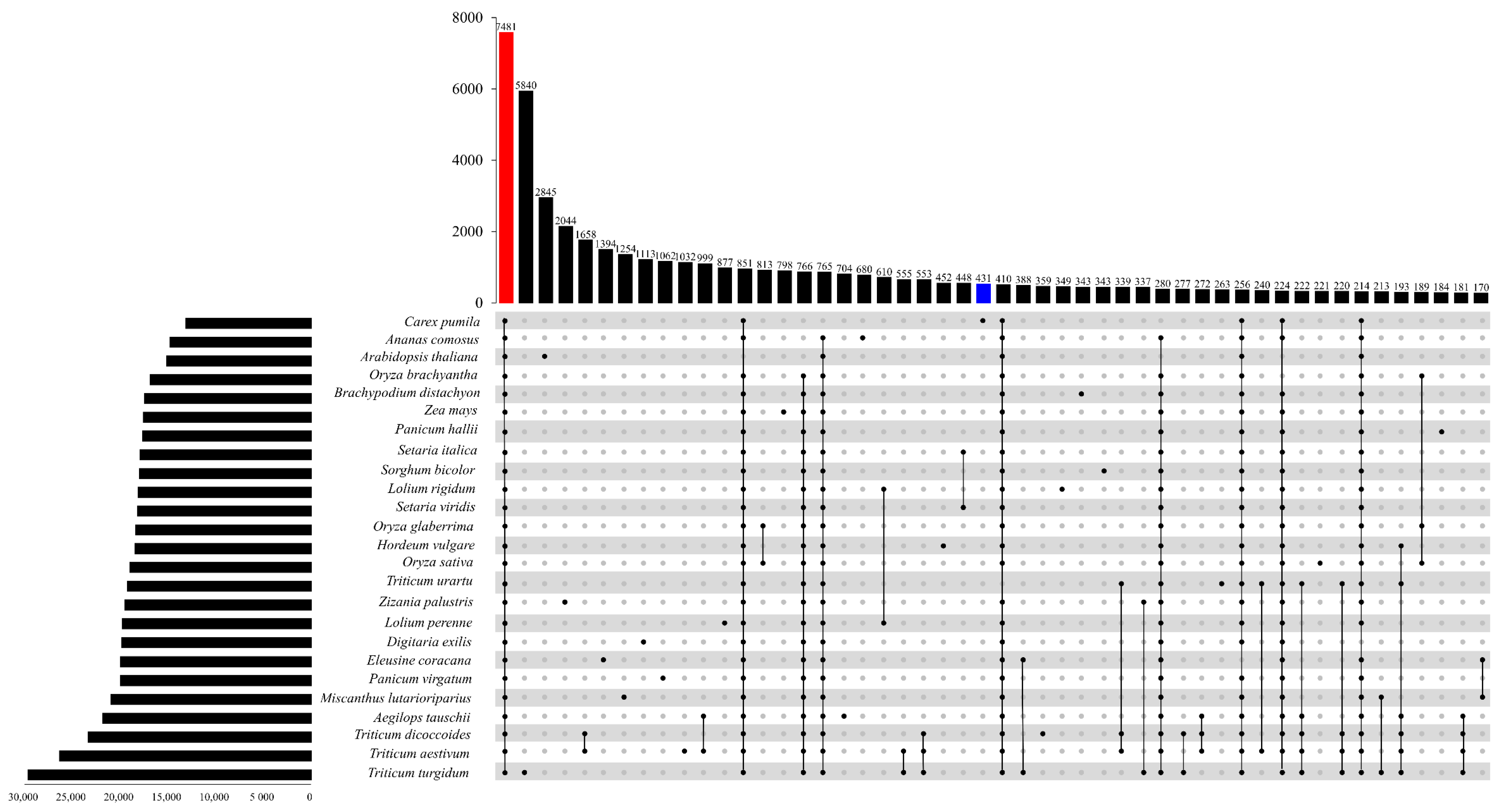
3.3. Phylogenetic Inference Orthologous Groups of C. pumila
3.4. Development of Novel Microsatellite Markers for Population Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bezerra, J.J.L.; Pinheiro, A.A.V. Traditional uses, phytochemistry, and anticancer potential of Cyperus rotundus L. (Cyperaceae): A systematic review. S. Afr. J. Bot. 2022, 144, 175–186. [Google Scholar] [CrossRef]
- Dávid, C.Z.; Hohmann, J.; Vasas, A. Chemistry and pharmacology of Cyperaceae stilbenoids: A review. Molecules 2021, 26, 2794. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Gupta, N.; Ghosh, R.; Gaonkar, R.H.; Pal, B.C. α-Glucosidase and α-amylase inhibitory constituent of Carex baccans: Bio-assay guided isolation and quantification by validated RP-HPLC–DAD. J. Funct. Foods 2013, 5, 211–218. [Google Scholar] [CrossRef]
- Simpson, D.A.; Inglis, C.A. Cyperaceae of economic, ethnobotanical and horticultural importance: A checklist. Kew Bull. 2001, 56, 257–360. [Google Scholar] [CrossRef]
- Lim, H.; Kim, Y.M. Carex pumila extract suppresses mast cell activation and IgE-mediated allergic response in mice. J. Food Saf. Hyg. 2014, 29, 356–362. [Google Scholar] [CrossRef]
- Ksouri, R.; Ksouri, W.M.; Jallali, I.; Debez, A.; Magné, C.; Hiroko, I.; Abdelly, C. Medicinal halophytes: Potent source of health promoting biomolecules with medical, nutraceutical and food applications. Crit. Rev. Biotechnol. 2012, 32, 289–326. [Google Scholar] [CrossRef]
- Kim, J.; Kong, C.S.; Seo, Y. Inhibitory effects of Carex pumila extracts on MMP-2 and MMP-9 activities in HT-1080 cells. Ocean Polar Res. 2018, 40, 249–257. [Google Scholar]
- Zhao, Q.; Yang, J.; Cui, M.Y.; Liu, J.; Fang, Y.; Yan, M.; Qui, W.; Shang, H.; Xu, Z.; Yidiresi, R.; et al. The reference genome sequence of Scutellaria baicalensis provides insights into the evolution of wogonin biosynthesis. Mol. Plant 2019, 12, 935–950. [Google Scholar] [CrossRef]
- Chakraborty, P. Development of new anti-diabetic drug from medicinal plant-genomic research. J Diabetol. 2018, 2, 6–7. [Google Scholar]
- Can, M.; Wei, W.; Zi, H.; Bai, M.; Liu, Y.; Gao, D.; Qu, G. Genome sequence of Kobresia littledalei, the first chromosome-level genome in the family Cyperaceae. Sci. Data 2020, 7, 175. [Google Scholar] [CrossRef]
- Ning, Y.; Li, Y.; Dong, S.B.; Yang, H.G.; Li, C.Y.; Xiong, B.; Yang, J.; Hu, Y.K.; Mu, X.Y.; Xia, X.F. The chromosome-scale genome of Kobresia myosuroides sheds light on karyotype evolution and recent diversification of a dominant herb group on the Qinghai-Tibet Plateau. DNA Res. 2023, 30, dsac049. [Google Scholar] [CrossRef]
- Qu, G.; Bao, Y.; Liao, Y.; Liu, C.; Zi, H.; Bai, M.; Lui, Y.; Tu, D.; Wang, L.; Chen, S.; et al. Draft genomes assembly and annotation of Carex parvula and Carex kokanica reveals stress-specific genes. Sci. Rep. 2022, 12, 4970. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yi, L.; Ren, Y.; Li, J.; Ren, W.; Hou, Z.; Su, S.; Wang, J.; Zhang, Y.; Dong, Q.; et al. Chromosome-scale genome assembly of the yellow nutsedge (Cyperus esculentus). Genome Biol. Evol. 2023, 15, evad027. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Lin, F.; An, D.; Wang, W.; Huang, R. Genome sequencing and assembly by long reads in plants. Genes 2017, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Jiao, W.B.; Schneeberger, K. The impact of third generation genomic technologies on plant genome assembly. Curr. Opin. Plant Biol. 2017, 36, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Hasan, N.; Choudhary, S.; Naaz, N.; Sharma, N.; Laskar, R.A. Recent advancements in molecular marker-assisted selection and applications in plant breeding programmes. J. Genet. Eng. Biotechnol. 2021, 19, 1–26. [Google Scholar] [CrossRef]
- Barrett, M.D.; Wallace, M.J.; Anthony, J.M. Characterization and cross application of novel microsatellite markers for a rare sedge, Lepidosperma gibsonii (Cyperaceae). Am. J. Bot. 2012, 99, e14–e16. [Google Scholar] [CrossRef]
- Blum, M.J.; Mclachlan, J.S.; Saunders, C.J.; Herrick, J.D. Characterization of microsatellite loci in Schoenoplectus americanus (Cyperaceae). Mol. Ecol. Notes 2022, 5, 661–663. [Google Scholar] [CrossRef]
- Böckelmann, J.; Wieser, D.; Tremetsberger, K.; Šumberová, K.; Bernhardt, K.G. Isolation of nuclear microsatellite markers for Cyperus fuscus (Cyperaceae). Appl. Plant Sci. 2015, 3, 1500071. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.J.; Mackay, D.A.; Whalen, M.A. Isolation of microsatellites from Baumea juncea (Cyperaceae). Conserv. Genet. Resour. 2011, 3, 113–115. [Google Scholar] [CrossRef]
- Gillespie, E.L.; Pauley, A.G.; Haffner, M.L.; Hay, N.M.; Estep, M.C.; Murrell, Z.E. Fourteen polymorphic microsatellite markers for a widespread limestone endemic, Carex eburnea (Cyperaceae: Carex sect. Albae). Appl. Plant Sci. 2017, 5, 1700031. [Google Scholar] [CrossRef]
- Hipp, A.L.; Kettenring, K.M.; Feldheim, K.A.; Weber, J.A. Isolation of 11 polymorphic tri-and tetranucleotide microsatellite loci in a North American sedge (Carex scoparia: Cyperaceae) and cross-species amplification in three additional Carex species. Mol. Ecol. Resour. 2009, 9, 625–627. [Google Scholar] [CrossRef] [PubMed]
- Ohsako, T.; Yamane, K. Isolation and characterization of polymorphic microsatellite loci in Asiatic sand sedge, Carex kobomugi Ohwi (Cyperaceae). Mol. Ecol. Notes 2007, 7, 1023–1025. [Google Scholar] [CrossRef]
- King, M.G.; Roalson, E.H. Isolation and characterization of 11 microsatellite loci from Carex macrocephala (Cyperaceae). Conserv. Genet. 2009, 10, 531–533. [Google Scholar] [CrossRef]
- Șuteu, D.; Pușcaș, M.; Băcilă, I.; Miclăuș, M.; Balázs, Z.R.; Choler, P. Development of SSR markers for Carex curvula (Cyperaceae) and their importance in investigating the species genetic structure. Mol. Biol. Rep. 2023, 50, 4729–4733. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.; Li, H. Fast and accurate long-read assembly with wtdbg2. Nat. Methods 2020, 17, 155–158. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H. Minimap and miniasm: Fast mapping and de novo assembly for noisy long sequences. Bioinformatics 2016, 32, 2103–2110. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Marçais, G.; Kingsford, C. Fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Vurture, G.W.; Sedlazeck, F.J.; Nattestad, M.; Underwood, C.J.; Fang, H.; Gurtowski, J.; Schatz, M.C. GenomeScope: Fast reference-free genome profiling from short reads. Bioinformatics 2017, 33, 2202–2204. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.S.; Holt, C.; Moore, B.; Yandell, M. Genome annotation and curation using MAKER and MAKER-P. Curr. Protoc. Bioinform 2014, 48, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Moriya, Y.; Itoh, M.; Okuda, S.; Kanehisa, M. KAAS: KEGG automatic annotation server. Genome Inform. 2005, 5. [Google Scholar]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef]
- Mendes, F.K.; Vanderpool, D.; Fulton, B.; Hahn, M.W. CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics 2020, 36, 5516–5518. [Google Scholar] [CrossRef]
- Schuelke, M. An economic method for the fluorescent labeling of PCR fragments. Nat. Biotechnol. 2000, 18, 233–234. [Google Scholar] [CrossRef]
- Kalinowski, S.T.; Taper, M.L.; Marshall, T.C. Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 2007, 16, 1099–1106. [Google Scholar] [CrossRef]
- Rojas-Gómez, M.; Jiménez-Madrigal, J.P.; Montero-Vargas, M.; Loaiza-Montoya, R.; Chavarría, M.; Meneses, E.; Fuchs, E.J. A draft genome assembly of “Cas” (Psidium friedrichsthalianum (O. Berg) Nied.): An indigenous crop of Costa Rica untapped. Genet. Resour. Crop Evol. 2022, 69, 39–47. [Google Scholar] [CrossRef]
- Vaziriyeganeh, M.; Khan, S.; Zwiazek, J.J. Transcriptome and metabolome analyses reveal potential salt tolerance mechanisms contributing to maintenance of water balance by the halophytic grass Puccinellia nuttalliana. Front. Plant Sci. 2021, 12, 760863. [Google Scholar] [CrossRef]
- Tan, C.; Zhang, H.; Chen, H.; Guan, M.; Zhu, Z.; Cao, X.; Ge, X.; Zhu, B.; Chen, D. First Report on Development of Genome-Wide Microsatellite Markers for Stock (Matthiola incana L.). Plants 2023, 12, 748. [Google Scholar] [CrossRef]
- Xu, J.; Liu, L.; Xu, Y.; Chen, C.; Rong, T.; Ali, F.; Zhou, S.; Wu, F.; Lui, Y.; Wang, J.; et al. Development and characterization of simple sequence repeat markers providing genome-wide coverage and high resolution in maize. DNA Res. 2013, 20, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Huang, K.; Nie, J.; Zhang, Y.; Zhang, Y.; Li, Y.; Wang, W.; Xu, X.; Ruan, R.; Yuan, X.; et al. Genome-wide mining, characterization, and development of microsatellite markers in Tartary buckwheat (Fagopyrum tataricum Garetn.). Euphytica 2019, 215, 1–10. [Google Scholar] [CrossRef]
- Katti, M.V.; Ranjekar, P.K.; Gupta, V.S. Differential distribution of simple sequence repeats in eukaryotic genome sequences. Mol. Biol. Evol. 2020, 18, 1161–1167. [Google Scholar] [CrossRef]
- Cavagnaro, P.F.; Senalik, D.A.; Yang, L.; Simon, P.W.; Harkins, T.T.; Kodira, C.D.; Huang, S.; Weng, Y. Genome-wide characterization of simple sequence repeats in cucumber (Cucumis sativus L.). BMC Genom. 2010, 11, 569. [Google Scholar] [CrossRef] [PubMed]
- Prinz, K.; Schie, S.; Debener, T.; Hensen, I.; Weising, K. Microsatellite markers for Spergularia media (L.) C. Presl. (Caryophyllaceae) and their cross-species transferability. Mol. Ecol. Resour. 2009, 9, 1424–1426. [Google Scholar] [CrossRef] [PubMed]
- Hodoki, Y.; Ohbayashi, K.; Kunii, H. Genetic analysis of salt-marsh sedge Carex scabrifolia Steud. populations using newly developed microsatellite markers. Conserv. Genet. 2009, 10, 1361–1364. [Google Scholar] [CrossRef]
- Korbecka-Glinka, G.; Skomra, U.; Olszak-Przybys, H. Cultivar identification in dry hop cones and pellets using microsatellite loci. Eur. Food Res. Technol. 2016, 242, 1599–1605. [Google Scholar] [CrossRef]
- Gómez-Rodríguez, M.V.; Beuzon, C.; González-Plaza, J.J.; Fernández-Ocaña, A.M. Identification of the olive (Olea europaea L.) core collection with a new set of SSR markers. Genet. Resour. Crop Evol. 2021, 68, 117–133. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Short Reads | Total Reads | Total Reads Length (bp) | Q20 (%) | Q30 (%) |
|---|---|---|---|---|
| Raw data | 870,146,384 | 131,392,103,984 | 92.31 | 83.52 |
| Filtered data | 350,433,302 | 52,800,516,376 | 99.47 | 97.17 |
| N50 (bp) | Mean subread length (bp) | |||
| Subreads data | 33,136,805 | 127,339,898,260 | 5273 | 3842 |
| Subreads (Iso-seq) | 18,257,235 | 24,044,828,716 | 1462 | 1317 |
| Assembler | HGAP | Wtdbg2 |
|---|---|---|
| Contig | 2941 | 4385 |
| Total contig bases (bp) | 346,579,715 | 338,223,786 |
| N50 (bp) | 351,713 | 160,342 |
| Max. length (bp) | 1,780,279 | 1,030,000 |
| Average length (bp) | 117,844 | 77,131 |
| Min. length (bp) | 1345 | 1661 |
| GC contents (%) | 33.4 | 33.9 |
| Status | HGAP | Wtdbg2 |
|---|---|---|
| Complete BUSCO (C) | 1481 (91.76%) | 1390 (86.12%) |
| Complete and single-copy BUSCO (S) | 1416 (87.73%) | 1367 (84.70%) |
| Complete and duplicated BUSCO (D) | 65 (4.02%) | 23 (1.43%) |
| Fragmented BUSCO (F) | 31 (1.92%) | 76 (4.71%) |
| Missing BUSCO (M) | 102 (6.32%) | 148 (9.17%) |
| Total BUSCO groups searched | 1614 (100%) | 1614 (100%) |
| Numbers of Genes | Length (bp) and Match Gene (%) | |
|---|---|---|
| Predicted proteins | 23,402 | 35,450,136 bp |
| EggNOG | 22,464 | 95.99 |
| InterPro | 18,762 | 80.17 |
| Pfam | 19,190 | 82.00 |
| COG | 9741 | 41.62 |
| KEGG | 8667 | 37.04 |
| Result of Microsatellites Search | |
|---|---|
| Total number of contigs sequences examined | 2941 |
| Total size of examined sequences (bp) | 346,579,715 |
| Total number of identified microsatellites | 62,565 |
| Number of microsatellites containing contigs sequences | 2328 |
| Di-nucleotides | 55,751 |
| Tri-nucleotides | 22,650 |
| Tetra-nucleotides | 6970 |
| Penta-nucleotides | 3628 |
| Hexa-nucleotides | 3162 |
| Locus | GenBank Accession No. | Primer Sequences (5′−3′) | Repeat Motif | Annealing Tm (°C) | Fluorescent Labels | Product Size | N | NA | HO | HE | PIC |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CapuMS10 | OQ685862 | F: TGTAAAACGACGGCCAGTTGTTTATGGAAGCTTCTCCTGGT R: CGGTTCCGATCGACGTTTTG | AT(42) | 57 | FAM | 373 | 24 | 6 | 0.292 | 0.688 | 0.617 |
| CapuMS12 | OQ685863 | F: TGTAAAACGACGGCCAGTGGATGGACACCCCTCAACAA R: CCAACAAAAGACGCTCAGCA | AT(40) | 57 | VIC | 278 | 24 | 4 | 0.750 | 0.646 | 0.586 |
| CapuMS14 | OQ685864 | F: TGTAAAACGACGGCCAGTTCAATGGTCTGGAAACAACGA R: CGGTTTGGATCGACAGTTTGG | TA(39) | 57 | FAM | 374 | 24 | 4 | 0.375 | 0.680 | 0.604 |
| CapuMS21 | OQ685865 | F: TGTAAAACGACGGCCAGTTCCAATGAAACCAGGTGCCT R: GCACAGGACCTCAGGAACAA | TA(38) | 57 | VIC | 286 | 24 | 8 | 0.250 | 0.741 | 0.691 |
| CapuMS23 | OQ685866 | F: TGTAAAACGACGGCCAGTCGCACTGCACGACTTTTGAA R: AACGGTTCGGATCGACTGTT | AT(36) | 57 | VIC | 267 | 24 | 4 | 0.958 | 0.666 | 0.585 |
| CapuMS27 | OQ685867 | F: TGTAAAACGACGGCCAGTTGGAGACATGATGGAGGCAT R: CAAGCCACTGGGGATTCTCA | TA(32) | 57 | FAM | 352 | 24 | 8 | 0.125 | 0.780 | 0.728 |
| CapuMS43 | OQ685868 | F: TGTAAAACGACGGCCAGTGCCGTAGCACGATAACAACC R: CCATAGCACCGTAGCCAAGT | AT(27) | 57 | NED | 324 | 24 | 4 | 0.250 | 0.622 | 0.560 |
| CapuMS45 | OQ685869 | F: TGTAAAACGACGGCCAGTTAGCACCCCAGCCTAACTCT R: TGTGATTTGGTTGCACCGTT | AT(26) | 57 | VIC | 265 | 24 | 5 | 0.375 | 0.712 | 0.654 |
| CapuMS48 | OQ685870 | F: TGTAAAACGACGGCCAGTTCCAGTTGCGAGTCACTTCC R: GTCGTAGCACCGTAGCTCTC | AT(24) | 57 | VIC | 210 | 24 | 6 | 0.542 | 0.829 | 0.785 |
| CapuMS56 | OQ685871 | F: TGTAAAACGACGGCCAGTTTGGACGGTGTACCTGCTTC R: TTCATTGGTTCGGTCCCTCT | TTC(30) | 57 | VIC | 211 | 24 | 12 | 0.875 | 0.895 | 0.864 |
| CapuMS57 | OQ685872 | F: TGTAAAACGACGGCCAGTTGTCTTCTTCATGCATGTTCGC R: AGAAATGGGGCTGTCTTCGT | AAT(30) | 57 | FAM | 374 | 24 | 6 | 0.458 | 0.559 | 0.516 |
| CapuMS59 | OQ685873 | F: TGTAAAACGACGGCCAGTGACGGCTATCGATCGAGCTT R: AGACAGTGACGTGATCCTCA | TTA(30) | 57 | PET | 153 | 24 | 9 | 0.458 | 0.847 | 0.808 |
| CapuMS61 | OQ685874 | F: TGTAAAACGACGGCCAGTCGGTCTCGCTCTACTTGCTT R: GCCATGTACCGGTGTCTGAT | TTA(28) | 57 | NED | 339 | 24 | 6 | 0.375 | 0.802 | 0.752 |
| CapuMS62 | OQ685875 | F: TGTAAAACGACGGCCAGTTGAGAGGTCAAAACGGAGTGA R: ATCCGATCCGCTACTGTGTC | TAT(27) | 57 | PET | 196 | 24 | 8 | 0.792 | 0.839 | 0.796 |
| CapuMS64 | OQ685876 | F: TGTAAAACGACGGCCAGTAGCAAGCTAGCCCTTTGACT R: AACGATTGGTACTCTGAGCA | TAT(26) | 57 | VIC | 214 | 24 | 4 | 0.375 | 0.630 | 0.554 |
| CapuMS66 | OQ685877 | F: TGTAAAACGACGGCCAGTCCAGAAGTGGGGTTTGATTTGG R: TGTCCAAGTGTCAGTTTCCAAC | ATA(26) | 57 | VIC | 316 | 24 | 8 | 0.792 | 0.856 | 0.818 |
| CapuMS68 | OQ685878 | F: TGTAAAACGACGGCCAGTAGCCGTAGTACCGTAGTCCT R: AAGGCTTTTCACCCTGTCAA | ATA(25) | 57 | VIC | 323 | 24 | 6 | 0.792 | 0.723 | 0.670 |
| CapuMS71 | OQ685879 | F: TGTAAAACGACGGCCAGTCACTTTCGGTTGGTGCGATT R: ATAAGCGCAGACCTGTGACA | TTA(24) | 57 | NED | 312 | 24 | 5 | 0.333 | 0.670 | 0.605 |
| CapuMS73 | OQ685880 | F: TGTAAAACGACGGCCAGTTCGGAGTCCCTCTCTTCCTT R: ACACCATTGTCATACTAGCCAGA | TAT(22) | 57 | NED | 304 | 24 | 7 | 0.667 | 0.696 | 0.644 |
| CapuMS76 | OQ685881 | F: TGTAAAACGACGGCCAGTAGCCAGCAAAACTTAATCACGA R: TTTCAATCTCGGCCGTTGGA | ATA(21) | 57 | PET | 178 | 24 | 5 | 0.833 | 0.752 | 0.691 |
| CapuMS77 | OQ685882 | F: TGTAAAACGACGGCCAGTAAACAGTCGTGTCACCATCT R: GGACAGATCCGGACACCATT | ATA(20) | 57 | NED | 328 | 24 | 5 | 0.125 | 0.623 | 0.544 |
| CapuMS84 | OQ685883 | F: TGTAAAACGACGGCCAGTCGGAGGACAAGATGAACGGT R: CGGGAATAACACCGTCTGCTA | TCTA(10) | 57 | FAM | 363 | 24 | 4 | 0.375 | 0.719 | 0.647 |
| CapuMS85 | OQ685884 | F: TGTAAAACGACGGCCAGTTCTGAGCTGTACGCATTCCC R: TTGTAGCACCGTAGCCCTTT | ATAC(10) | 57 | FAM | 353 | 24 | 5 | 0.750 | 0.719 | 0.653 |
| CapuMS88 | OQ685885 | F: TGTAAAACGACGGCCAGTCCGAGTCATGTGCACCACTA R: TCTGAACGGGGCAAGTATGT | TTAT(9) | 57 | NED | 303 | 24 | 5 | 0.333 | 0.773 | 0.716 |
| CapuMS89 | OQ685886 | F: TGTAAAACGACGGCCAGTTGCCACCTGTACGTGTAGTG R: GCACCGCTGGACACTACATA | AAAT(9) | 57 | NED | 332 | 24 | 4 | 0.792 | 0.554 | 0.474 |
| CapuMS90 | OQ685887 | F: TGTAAAACGACGGCCAGTCACCACGGTTCCGAAACAAA R: AGCAACGTCTACCATTGGCA | ACAT(9) | 57 | PET | 180 | 24 | 4 | 0.583 | 0.699 | 0.633 |
| CapuMS95 | OQ685888 | F: TGTAAAACGACGGCCAGTCACCCTCCAATCCATCACGA R: GACCGACATCGAGTGAAGGA | CTGGG(10) | 57 | FAM | 371 | 24 | 6 | 0.250 | 0.730 | 0.669 |
| CapuMS96 | OQ685889 | F: TGTAAAACGACGGCCAGTCTGGGCTGATGTCAGGTTGT R: CCACACGCGTGCATTTAATCT | GTATT(8) | 57 | VIC | 291 | 24 | 5 | 0.708 | 0.708 | 0.640 |
| CapuMS97 | OQ685890 | F: TGTAAAACGACGGCCAGTGGTCACTCATGTCTCCGTCA R: CGGAGGCAAAGCTTGAACAA | ACACC(8) | 57 | VIC | 237 | 24 | 4 | 0.583 | 0.720 | 0.651 |
| CapuMS99 | OQ685891 | F: TGTAAAACGACGGCCAGTTGTGGGTGCATCAGAGACAC R: TGGGACGTCTAGGGGACAAT | CATGC(8) | 57 | VIC | 287 | 24 | 4 | 0.125 | 0.708 | 0.637 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, K.-R.; Yu, J.-N.; Hong, J.M.; Kim, S.-Y.; Park, S.Y. Genome Assembly and Microsatellite Marker Development Using Illumina and PacBio Sequencing in the Carex pumila (Cyperaceae) from Korea. Genes 2023, 14, 2063. https://doi.org/10.3390/genes14112063
Kim K-R, Yu J-N, Hong JM, Kim S-Y, Park SY. Genome Assembly and Microsatellite Marker Development Using Illumina and PacBio Sequencing in the Carex pumila (Cyperaceae) from Korea. Genes. 2023; 14(11):2063. https://doi.org/10.3390/genes14112063
Chicago/Turabian StyleKim, Kang-Rae, Jeong-Nam Yu, Jeong Min Hong, Sun-Yu Kim, and So Young Park. 2023. "Genome Assembly and Microsatellite Marker Development Using Illumina and PacBio Sequencing in the Carex pumila (Cyperaceae) from Korea" Genes 14, no. 11: 2063. https://doi.org/10.3390/genes14112063