SNP Array Screening and Long Range PCR-Based Targeted Next Generation Sequencing for Autosomal Recessive Disease with Consanguinity: Insight from a Case of Xeroderma Pigmentosum Group C



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Case Presentation
2.2. Methods of Molecular Diagnosis
2.2.1. Mutation Analysis
2.2.2. Immunohistochemical Staining of XPC Protein
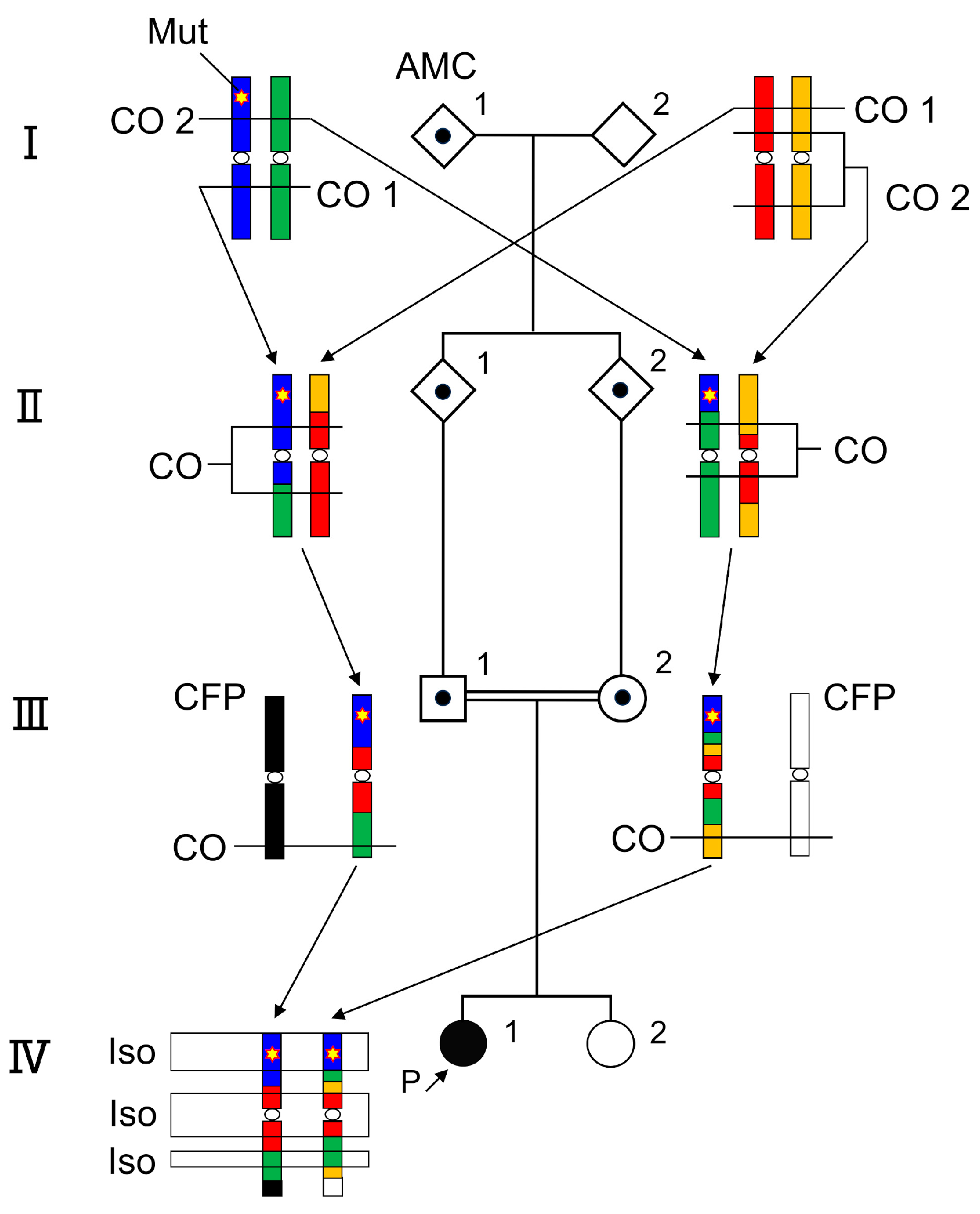
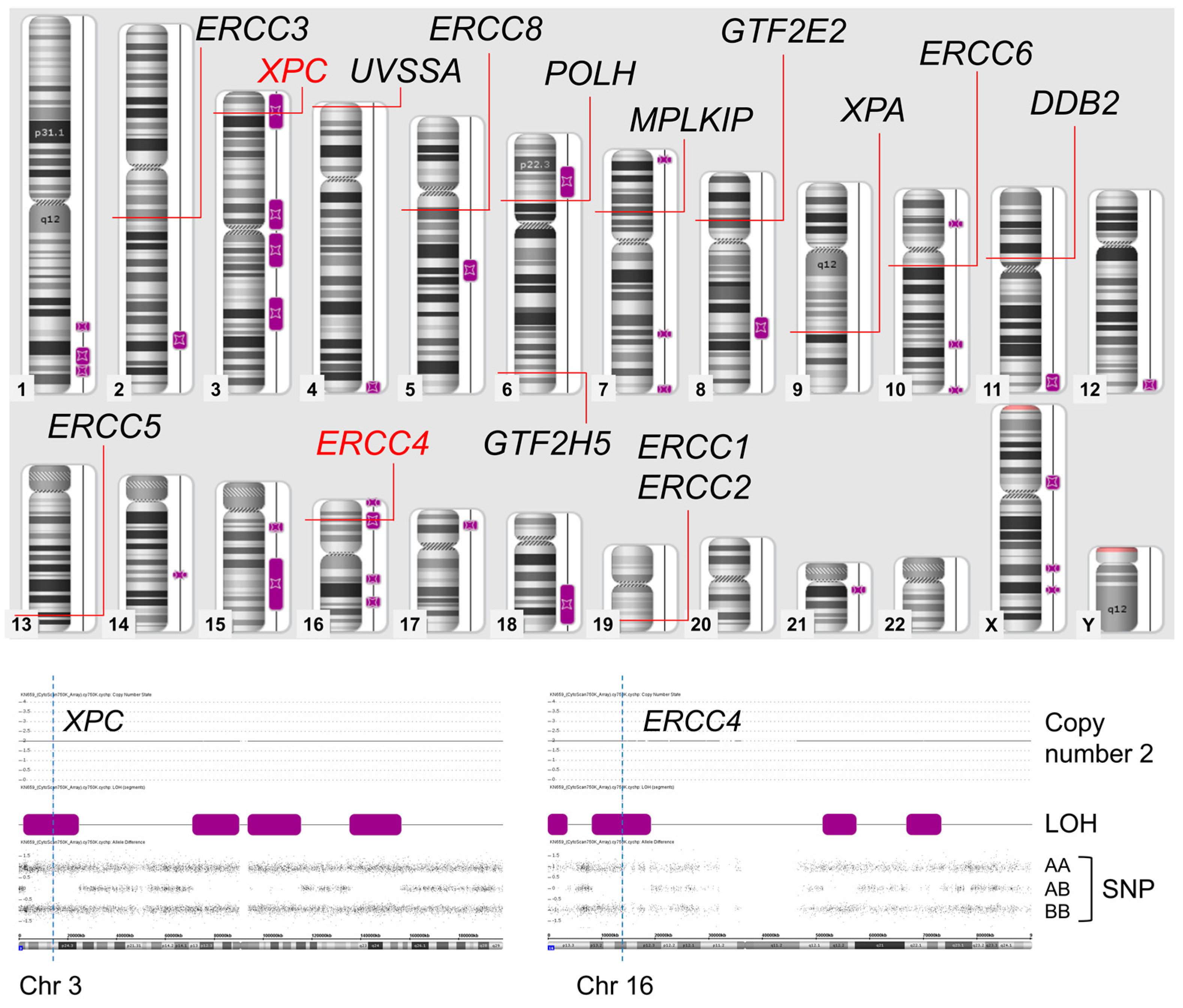
3. Results
3.1. SNP Array
3.2. Long Range PCR-Based NGS and Validation of Sanger Sequencing
3.3. Immunohistochemistry
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taylor, A.; Alloub, Z.; Tayoun, A.A. A Simple Practical Guide to Genomic Diagnostics in a Pediatric Setting. Genes 2021, 12, 818. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shen, X.; Fang, F.; Ding, C.H.; Zhang, H.; Cao, Z.H.; An, D.Y. Phenotype-Driven Virtual Panel Is an Effective Method to Analyze WES Data of Neurological Disease. Front. Pharmacol. 2018, 9, 1529. [Google Scholar] [CrossRef] [PubMed]
- Fledel-Alon, A.; Wilson, D.J.; Broman, K.; Wen, X.; Ober, C.; Coop, G.; Przeworski, M. Broad-scale recombination patterns underlying proper disjunction in humans. PLoS Genet. 2009, 5, e1000658. [Google Scholar] [CrossRef]
- Sarrate, Z.; Vidal, F.; Blanco, J. Meiotic abnormalities in metaphase I human spermatocytes from infertile males: Frequencies, chromosomes involved, and the relationships with polymorphic karyotype and seminal parameters. Asian J. Androl. 2014, 16, 838–844. [Google Scholar] [CrossRef] [PubMed]
- Togi, S.; Ura, H.; Niida, Y. Optimization and Validation of Multimodular, Long-Range PCR-Based Next-Generation Sequencing Assays for Comprehensive Detection of Mutation in Tuberous Sclerosis Complex. J. Mol. Diagn. 2021, 23, 424–446. [Google Scholar] [CrossRef]
- Togi, S.; Ura, H.; Niida, Y. Application of Combined Long Amplicon Sequencing (CoLAS) for Genetic Analysis of Neurofibromatosis Type 1: A Pilot Study. Curr. Issues Mol. Biol. 2021, 43, 782–801. [Google Scholar] [CrossRef] [PubMed]
- Adam, M.P.; Mirzaa, G.M.; Pagon, R.A.; Wallace, S.E.; Bean, L.J.H.; Gripp, K.W.; Amemiya, A. GeneReviews. In 1993. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1397/ (accessed on 20 October 2023).
- Leung, A.K.; Barankin, B.; Lam, J.M.; Leong, K.F.; Hon, K.L. Xeroderma pigmentosum: An updated review. Drugs Context 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Brambullo, T.; Colonna, M.R.; Vindigni, V.; Piaserico, S.; Masciopinto, G.; Galeano, M.; Costa, A.L.; Bassetto, F. Xeroderma Pigmentosum: A Genetic Condition Skin Cancer Correlated—A Systematic Review. BioMed Res. Int. 2022, 2022, 8549532. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, D.K.; Schnabel, B. DNA isolation by a rapid method from human blood samples: Effects of MgCl2, EDTA, storage time, and temperature on DNA yield and quality. Biochem. Genet. 1993, 31, 321–328. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wangle, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Supek, F.; Lehner, B.; Lindeboom, R.G.H. To NMD or Not To NMD: Nonsense-Mediated mRNA Decay in Cancer and Other Genetic Diseases. Trends Genet. 2021, 37, 657–668. [Google Scholar] [CrossRef]
- Zhang, E.T.; He, Y.; Grob, P.; Fong, Y.W.; Nogales, E.; Tjian, R. Architecture of the human XPC DNA repair and stem cell coactivator complex. Proc. Natl. Acad. Sci. USA 2015, 112, 14817–14822. [Google Scholar] [CrossRef]
- Bellus, G.A.; Hefferon, T.W.; Ortiz de Luna, R.I.; Hecht, J.T.; Horton, W.A.; Machado, M.; Kaitila, I.; McIntosh, I.; Francomano, C.A. Achondroplasia is defined by recurrent G380R mutations of FGFR3. Am. J. Hum. Genet. 1995, 56, 368–373. [Google Scholar]
- Pauli, R.M. Achondroplasia: A comprehensive clinical review. Orphanet J. Rare Dis. 2019, 14, 1. [Google Scholar] [CrossRef]
- Feliubadaló, L.; Lopez-Doriga, A.; Castellsagué, E.; del Valle, J.; Menéndez, M.; Tornero, E.; Montes, E.; Cuesta, R.; Gómez, C.; Campos, O.; et al. Next-generation sequencing meets genetic diagnostics: Development of a comprehensive workflow for the analysis of BRCA1 and BRCA2 genes. Eur. J. Hum. Genet. 2013, 21, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Pasmant, E.; Parfait, B.; Luscan, A.; Goussard, P.; Briand-Suleau, A.; Laurendeau, I.; Fouveaut, C.; Leroy, C.; Montadert, A.; Wolkenstein, P.; et al. Neurofibromatosis type 1 molecular diagnosis: What can NGS do for you when you have a large gene with loss of function mutations? Eur. J. Hum. Genet. 2015, 23, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Lappalainen, T.; Scott, A.J.; Brandt, M.; Hall, I.M. Genomic Analysis in the Age of Human Genome Sequencing. Cell 2019, 177, 70–84. [Google Scholar] [CrossRef]
- Reuter, C.M.; Kohler, J.N.; Bonner, D.; Zastrow, D.; Fernandez, L.; Dries, A.; Marwaha, S.; Davidson, J.; Brokamp, E.; Herzog, M.; et al. Yield of whole exome sequencing in undiagnosed patients facing insurance coverage barriers to genetic testing. J. Genet. Couns. 2019, 28, 1107–1118. [Google Scholar] [CrossRef]
- Takahashi, Y.; Mizusawa, H. Initiative on Rare and Undiagnosed Disease in Japan. JMA J. 2021, 4, 112–118. [Google Scholar] [PubMed]
- Abdellaoui, A.; Yengo, L.; Verweij, K.J.H.; Visscher, P.M. 15 years of GWAS discovery: Realizing the promise. Am. J. Hum. Genet. 2023, 110, 179–194. [Google Scholar] [CrossRef]
- Ciani, M.; Benussi, L.; Bonvicini, C.; Ghidoni, R. Genome Wide Association Study and Next Generation Sequencing: A Glimmer of Light Toward New Possible Horizons in Frontotemporal Dementia Research. Front. Neurosci. 2019, 13, 506. [Google Scholar] [CrossRef]
- den Hollander, A.I.; Lopez, I.; Yzer, S.; Zonneveld, M.N.; Janssen, I.M.; Strom, T.M.; Hehir-Kwa, J.Y.; Veltman, J.A.; Arends, M.L.; Meitinger, T.; et al. Identification of novel mutations in patients with Leber congenital amaurosis and juvenile RP by genome-wide homozygosity mapping with SNP microarrays. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5690–5698. [Google Scholar] [CrossRef]
- Chiang, A.P.; Beck, J.S.; Yen, H.J.; Tayeh, M.K.; Scheetz, T.E.; Swiderski, R.E.; Nishimura, D.Y.; Braun, T.A.; Kim, K.Y.; Huang, J.; et al. Homozygosity mapping with SNP arrays identifies TRIM32, an E3 ubiquitin ligase, as a Bardet-Biedl syndrome gene (BBS11). Proc. Natl. Acad. Sci. USA 2006, 103, 6287–6292. [Google Scholar] [CrossRef]
- Santoro, S.L.; Hashimoto, S.; McKinney, A.; Mihalic Mosher, T.; Pyatt, R.; Reshmi, S.C.; Astbury, C.; Hickey, S.E. Assessing the Clinical Utility of SNP Microarray for Prader-Willi Syndrome due to Uniparental Disomy. Cytogenet. Genome Res. 2017, 152, 105–109. [Google Scholar] [CrossRef]
- Izumi, K.; Santani, A.B.; Deardorff, M.A.; Feret, H.A.; Tischler, T.; Thiel, B.D.; Mulchandani, S.; Stolle, C.A.; Spinner, N.B.; Zackai, E.H.; et al. Mosaic maternal uniparental disomy of chromosome 15 in Prader-Willi syndrome: Utility of genome-wide SNP array. Am. J. Med. Genet. A 2013, 161A, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Niida, Y.; Ozaki, M.; Shimizu, M.; Ueno, K.; Tanaka, T. Classification of Uniparental Isodisomy Patterns That Cause Autosomal Recessive Disorders: Proposed Mechanisms of Different Proportions and Parental Origin in Each Pattern. Cytogenet. Genome Res. 2018, 154, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Papić, L.; Fischer, D.; Trajanoski, S.; Höftberger, R.; Fischer, C.; Ströbel, T.; Schmidt, W.M.; Bittner, R.E.; Schabhüttl, M.; Gruber, K.; et al. SNP-array based whole genome homozygosity mapping: A quick and powerful tool to achieve an accurate diagnosis in LGMD2 patients. Eur. J. Med. Genet. 2011, 54, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Fischer, C.; Trajanoski, S.; Papić, L.; Windpassinger, C.; Bernert, G.; Freilinger, M.; Schabhüttl, M.; Arslan-Kirchner, M.; Javaher-Haghighi, P.; Plecko, B.; et al. SNP array-based whole genome homozygosity mapping as the first step to a molecular diagnosis in patients with Charcot-Marie-Tooth disease. J. Neurol. 2012, 259, 515–523. [Google Scholar] [CrossRef]
- Ben Rekaya, M.; Laroussi, N.; Messaoud, O.; Jones, M.; Jerbi, M.; Naouali, C.; Bouyacoub, Y.; Chargui, M.; Kefi, R.; Fazaa, B.; et al. A founder large deletion mutation in Xeroderma pigmentosum—Variant form in Tunisia: Implication for molecular diagnosis and therapy. BioMed Res. Int. 2014, 2014, 256245. [Google Scholar] [CrossRef]
- Alberts, B.; Wilson, J.H.; Hunt, T.; Heald, R.; Johnson, A.D.; Morgan, D.O.; Raff, M.C.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 7th ed.; International Student, Ed.; W.W. Norton & Company: New York, NY, USA, 2022. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nomura, F.; Shimizu, A.; Togi, S.; Ura, H.; Niida, Y. SNP Array Screening and Long Range PCR-Based Targeted Next Generation Sequencing for Autosomal Recessive Disease with Consanguinity: Insight from a Case of Xeroderma Pigmentosum Group C. Genes 2023, 14, 2079. https://doi.org/10.3390/genes14112079
Nomura F, Shimizu A, Togi S, Ura H, Niida Y. SNP Array Screening and Long Range PCR-Based Targeted Next Generation Sequencing for Autosomal Recessive Disease with Consanguinity: Insight from a Case of Xeroderma Pigmentosum Group C. Genes. 2023; 14(11):2079. https://doi.org/10.3390/genes14112079
Chicago/Turabian StyleNomura, Fumie, Akira Shimizu, Sumihito Togi, Hiroki Ura, and Yo Niida. 2023. "SNP Array Screening and Long Range PCR-Based Targeted Next Generation Sequencing for Autosomal Recessive Disease with Consanguinity: Insight from a Case of Xeroderma Pigmentosum Group C" Genes 14, no. 11: 2079. https://doi.org/10.3390/genes14112079