
Epigenetic Profiling of Type 2 Diabetes Mellitus: An Epigenome-Wide Association Study of DNA Methylation in the Korean Genome and Epidemiology Study


Abstract
:1. Introduction
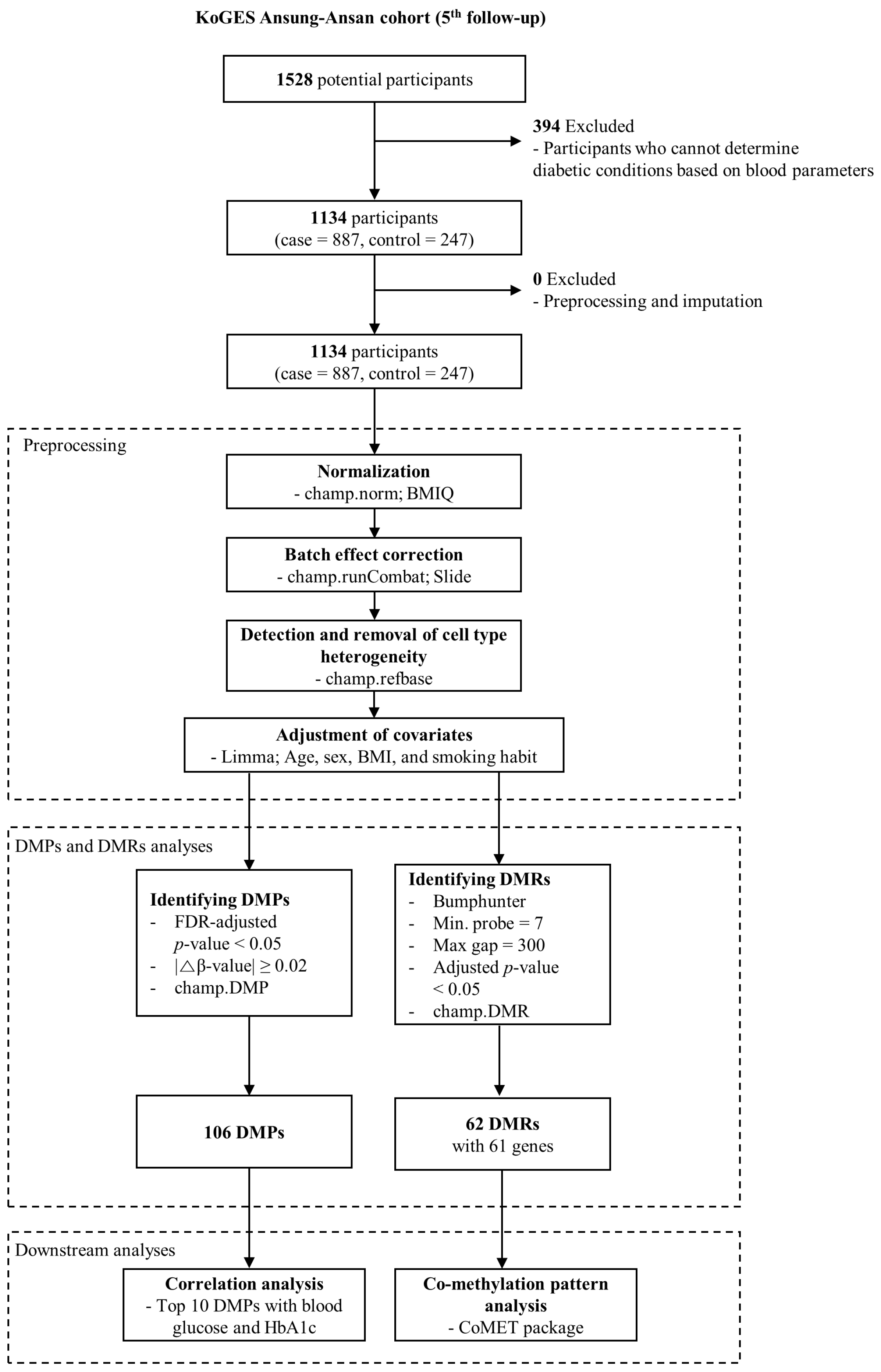
2. Materials and Methods
2.1. Participants and Data Source
2.2. Study Design
2.3. Data Preprocessing and Adjustment of Confounding Effects
2.4. Identification of DMPs
2.5. Identification of DMRs
2.6. Statistical Analyses
3. Results
3.1. Clinical Characteristics of the Study Participants
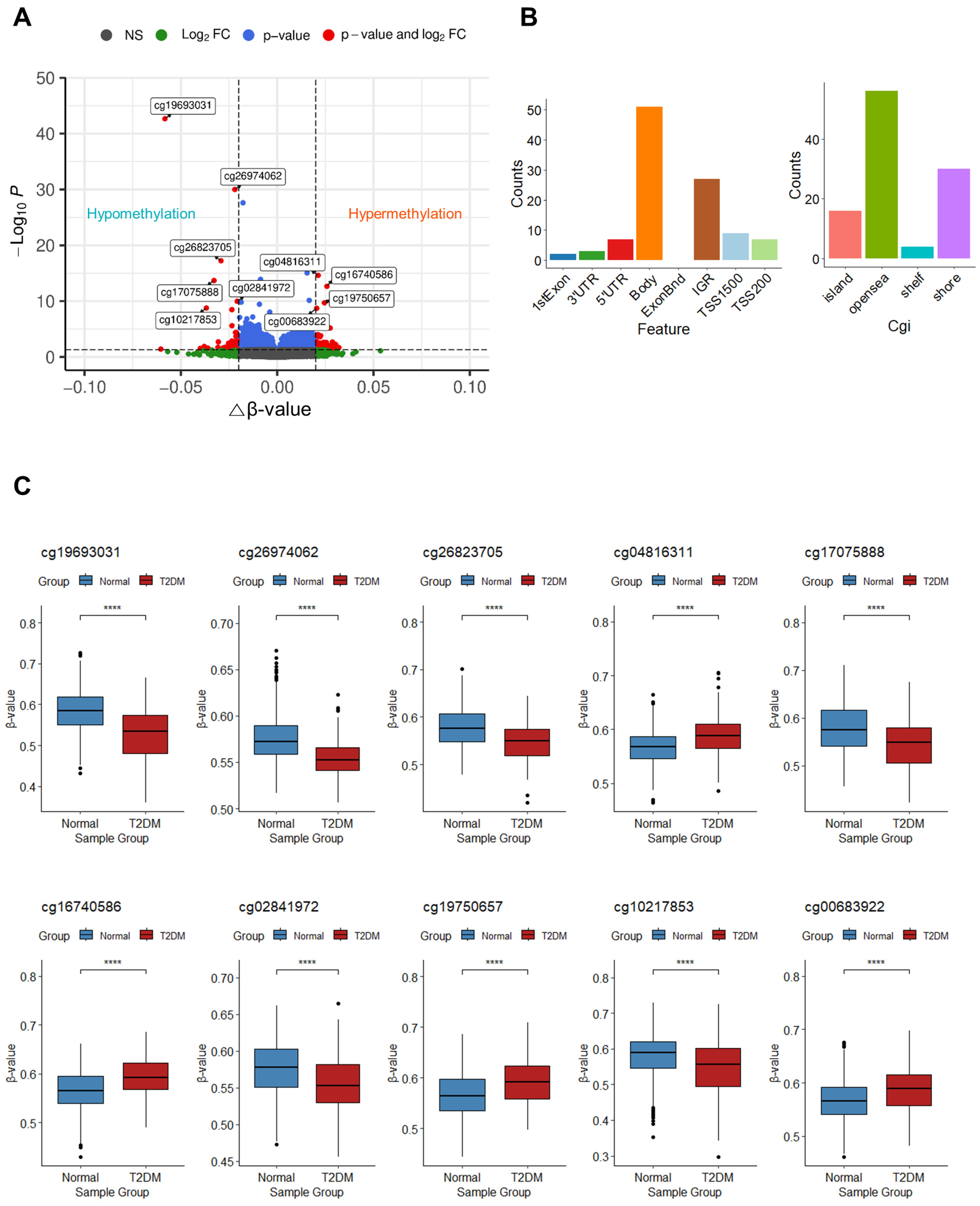
3.2. Identification of DMPs
3.3. Correlation between Methylation Levels of Top DMPs and Glycemic Markers (FPG and HbA1c)
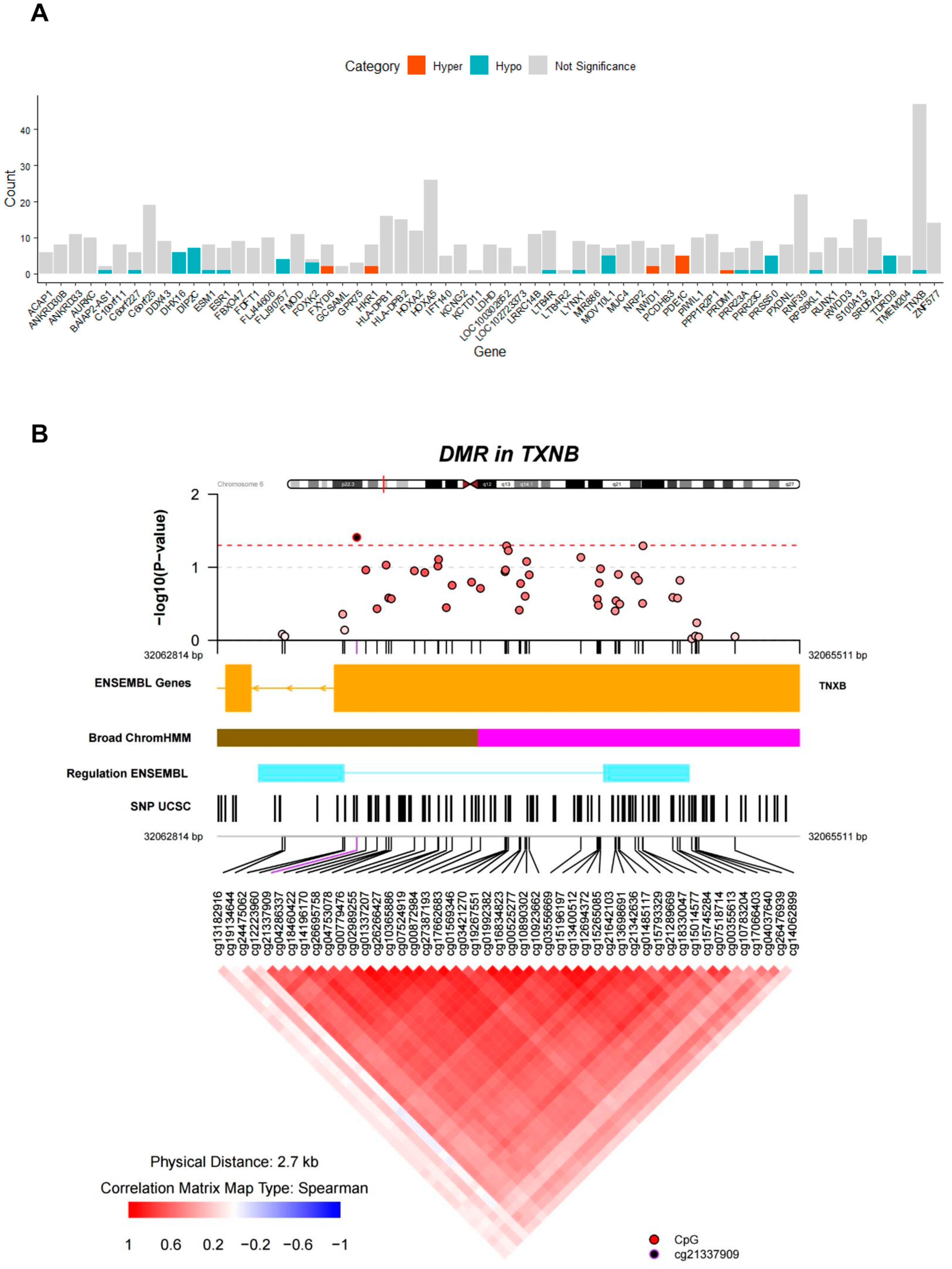
3.4. Identification of DMRs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- American Diabetes Association Professional Practice Committee. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes—2022. Diabetes Care 2022, 45, S17–S38. [Google Scholar] [CrossRef]
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.; Mbanya, J.C. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef]
- Zeng, P.; Zhao, Y.; Qian, C.; Zhang, L.; Zhang, R.; Gou, J.; Liu, J.; Liu, L.; Chen, F. Statistical analysis for genome-wide association study. J. Biomed. Res. 2015, 29, 285. [Google Scholar]
- Prasad, R.B.; Groop, L. Genetics of type 2 diabetes—Pitfalls and possibilities. Genes 2015, 6, 87–123. [Google Scholar] [CrossRef] [PubMed]
- Verma, M. Genome-wide association studies and epigenome-wide association studies go together in cancer control. Future Oncol. 2016, 12, 1645–1664. [Google Scholar] [CrossRef] [PubMed]
- Arpon, A.; Milagro, F.I.; Ramos-Lopez, O.; Mansego, M.L.; Santos, J.L.; Riezu-Boj, J.-I.; Martínez, J.A. Epigenome-wide association study in peripheral white blood cells involving insulin resistance. Sci. Rep. 2019, 9, 2445. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.A.; Zhang, E.; Natarajan, R. Epigenetic mechanisms in diabetic complications and metabolic memory. Diabetologia 2015, 58, 443–455. [Google Scholar] [CrossRef]
- Cardona, A.; Day, F.R.; Perry, J.R.; Loh, M.; Chu, A.Y.; Lehne, B.; Paul, D.S.; Lotta, L.A.; Stewart, I.D.; Kerrison, N.D. Epigenome-wide association study of incident type 2 diabetes in a British population: EPIC-Norfolk study. Diabetes 2019, 68, 2315–2326. [Google Scholar] [CrossRef] [PubMed]
- Juvinao-Quintero, D.L.; Marioni, R.E.; Ochoa-Rosales, C.; Russ, T.C.; Deary, I.J.; Van Meurs, J.B.; Voortman, T.; Hivert, M.-F.; Sharp, G.C.; Relton, C.L. DNA methylation of blood cells is associated with prevalent type 2 diabetes in a meta-analysis of four European cohorts. Clin. Epigenet. 2021, 13, 40. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Han, B.-G.; Group, K. Cohort profile: The Korean genome and epidemiology study (KoGES) consortium. Int. J. Epidemiol. 2017, 46, e20. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.K.; Kim, H.; Lee, Y.; Lee, Y.S.; Gim, J.A. DNA Methylation Patterns According to Fatty Liver Index and Longitudinal Changes from the Korean Genome and Epidemiology Study (KoGES). Curr. Issues Mol. Biol. 2022, 44, 1149–1168. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-E.; Jo, M.-J.; Cho, E.; Ahn, S.-Y.; Kwon, Y.-J.; Gim, J.-A.; Ko, G.-J. The Effect of DNA Methylation in the Development and Progression of Chronic Kidney Disease in the General Population: An Epigenome-Wide Association Study Using the Korean Genome and Epidemiology Study Database. Genes 2023, 14, 1489. [Google Scholar] [CrossRef]
- Martin, T.C.; Yet, I.; Tsai, P.-C.; Bell, J.T. coMET: Visualisation of regional epigenome-wide association scan results and DNA co-methylation patterns. BMC Bioinform. 2015, 16, 131. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Morris, T.J.; Webster, A.P.; Yang, Z.; Beck, S.; Feber, A.; Teschendorff, A.E. ChAMP: Updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics 2017, 33, 3982–3984. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; Marabita, F.; Lechner, M.; Bartlett, T.; Tegner, J.; Gomez-Cabrero, D.; Beck, S. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 2013, 29, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Blighe, K.; Rana, S.; Lewis, M. EnhancedVolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling; R Package Version; Bioconductor: Boston, MA, USA, 2019; Volume 1. [Google Scholar]
- Meeks, K.A.; Henneman, P.; Venema, A.; Addo, J.; Bahendeka, S.; Burr, T.; Danquah, I.; Galbete, C.; Mannens, M.M.; Mockenhaupt, F.P. Epigenome-wide association study in whole blood on type 2 diabetes among sub-Saharan African individuals: Findings from the RODAM study. Int. J. Epidemiol. 2019, 48, 58–70. [Google Scholar] [CrossRef]
- Chilunga, F.P.; Meeks, K.A.; Henneman, P.; Agyemang, C.; Doumatey, A.P.; Rotimi, C.N.; Adeyemo, A.A. An epigenome-wide association study of insulin resistance in African Americans. Clin. Epigenet. 2022, 14, 88. [Google Scholar] [CrossRef]
- Dawes, K.; Philibert, W.; Darbro, B.; Simons, R.L.; Philibert, R. Additive and interactive genetically contextual effects of HbA1c on cg19693031 methylation in type 2 diabetes. Genes 2022, 13, 683. [Google Scholar] [CrossRef] [PubMed]
- Mohammad Alhawiti, N.; Al Mahri, S.; Azhar Aziz, M.; Shafi Malik, S.; Mohammad, S. TXNIP in metabolic regulation: Physiological role and therapeutic outlook. Curr. Drug Targets 2017, 18, 1095–1103. [Google Scholar]
- Basnet, R.; Basnet, T.B.; Basnet, B.B.; Khadka, S. Overview on thioredoxin-interacting protein (TXNIP): A potential target for diabetes intervention. Curr. Drug Targets 2022, 23, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Thielen, L.; Shalev, A. Diabetes pathogenic mechanisms and potential new therapies based upon a novel target called TXNIP. Curr. Opin. Endocrinol. Diabetes Obes. 2018, 25, 75. [Google Scholar] [CrossRef]
- Moon, S.-S.; Lee, J.-E.; Lee, Y.-S.; Kim, S.-W.; Jeoung, N.H.; Lee, I.-K.; Kim, J.-G. Association of pyruvate dehydrogenase kinase 4 gene polymorphisms with type 2 diabetes and metabolic syndrome. Diabetes Res. Clin. Pract. 2012, 95, 230–236. [Google Scholar] [CrossRef]
- Sugden, M.; Holness, M. Therapeutic potential of the mammalian pyruvate dehydrogenase kinases in the prevention of hyperglycaemia. Curr. Drug Targets-Immune Endocr. Metab. Disord. 2002, 2, 151–165. [Google Scholar] [CrossRef]
- Lee, K.; Moon, S.; Park, M.-J.; Koh, I.-U.; Choi, N.-H.; Yu, H.-Y.; Kim, Y.J.; Kong, J.; Kang, H.G.; Kim, S.C. Integrated analysis of tissue-specific promoter methylation and gene expression profile in complex diseases. Int. J. Mol. Sci. 2020, 21, 5056. [Google Scholar] [CrossRef]
- Ling, C. Epigenetic regulation of insulin action and secretion–role in the pathogenesis of type 2 diabetes. J. Intern. Med. 2020, 288, 158–167. [Google Scholar] [CrossRef]
- Ochoa-Rosales, C.; Portilla-Fernandez, E.; Nano, J.; Wilson, R.; Lehne, B.; Mishra, P.P.; Gao, X.; Ghanbari, M.; Rueda-Ochoa, O.L.; Juvinao-Quintero, D. Epigenetic link between statin therapy and type 2 diabetes. Diabetes Care 2020, 43, 875–884. [Google Scholar] [CrossRef]
- Koh, I.-U.; Choi, N.-H.; Lee, K.; Yu, H.-Y.; Yun, J.H.; Kong, J.-H.; Kim, H.J.; Lee, S.; Kim, S.C.; Kim, B.-J. Obesity susceptible novel DNA methylation marker on regulatory region of inflammation gene: Results from the Korea Epigenome Study (KES). BMJ Open Diabetes Res. Care 2020, 8, e001338. [Google Scholar] [CrossRef]
- Kim, H.; Bae, J.H.; Park, K.S.; Sung, J.; Kwak, S.H. DNA methylation changes associated with type 2 diabetes and diabetic kidney disease in an East Asian population. J. Clin. Endocrinol. Metab. 2021, 106, e3837–e3851. [Google Scholar] [CrossRef] [PubMed]
- Yousri, N.A.; Albagha, O.M.; Hunt, S.C. Integrated epigenome, whole genome sequence and metabolome analyses identify novel multi-omics pathways in type 2 diabetes: A Middle Eastern study. BMC Med. 2023, 21, 347. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Yang, Y.; Allister, E.M.; Wijesekara, N.; Wheeler, M.B. The identification of potential factors associated with the development of type 2 diabetes: A quantitative proteomics approach. Mol. Cell. Proteom. 2008, 7, 1434–1451. [Google Scholar] [CrossRef] [PubMed]
- Richards, P.; Rachdi, L.; Oshima, M.; Marchetti, P.; Bugliani, M.; Armanet, M.; Postic, C.; Guilmeau, S.; Scharfmann, R. MondoA is an essential glucose-responsive transcription factor in human pancreatic β-cells. Diabetes 2018, 67, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Carruthers, N.J.; Strieder-Barboza, C.; Caruso, J.A.; Flesher, C.G.; Baker, N.A.; Kerk, S.A.; Ky, A.; Ehlers, A.P.; Varban, O.A.; Lyssiotis, C.A. The human type 2 diabetes-specific visceral adipose tissue proteome and transcriptome in obesity. Sci. Rep. 2021, 11, 17394. [Google Scholar] [CrossRef]
- Daniel, P.V.; Kamthan, M.; Thakur, S.; Mondal, P. Molecular pathways dysregulated by Pb2+ exposure prompts pancreatic beta-cell dysfunction. Toxicol. Res. 2022, 11, 206–214. [Google Scholar] [CrossRef]
- Niu, T.; Qin, Z.S.; Xu, X.; Liu, J.S. Bayesian haplotype inference for multiple linked single-nucleotide polymorphisms. Am. J. Hum. Genet. 2002, 70, 157–169. [Google Scholar] [CrossRef]
- Akbari, V.; Garant, J.-M.; O’Neill, K.; Pandoh, P.; Moore, R.; Marra, M.A.; Hirst, M.; Jones, S.J. Megabase-scale methylation phasing using nanopore long reads and NanoMethPhase. Genome Biol. 2021, 22, 68. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KoGES Cohort (n = 1134) | Normal (n = 887) | T2DM (n = 247) | p Value a | ||||
|---|---|---|---|---|---|---|---|
| Mean | SD | n | Mean | SD | n | ||
| Sex (% male) | 50.20 | 887 | 57.50 | 247 | - | ||
| Age (years) | 57.77 | 8.38 | 887 | 62.45 | 8.22 | 247 | *** |
| BMI (kg/m2) | 23.64 | 2.77 | 887 | 25.31 | 2.79 | 247 | *** |
| Smoking habit (%) | 37.80 | 887 | 47.30 | 247 | - | ||
| Fasting glucose (mg/dL) | 89.26 | 5.64 | 887 | 161.05 | 40.97 | 247 | *** |
| HbA1c (%) | 5.35 | 0.21 | 887 | 7.56 | 1.05 | 247 | *** |
| 2-h plasma glucose (mg/dL) | 105.49 | 21.25 | 887 | 261.96 | 58.38 | 56 | *** |
| Fasting insulin (µIU/mL) | 7.63 | 3.04 | 887 | 13.57 | 18.69 | 247 | *** |
| 2-h plasma insulin (µIU/mL) | 28.69 | 27.68 | 887 | 36.68 | 29.04 | 56 | *** |
| Newly detected DM (n, %) | 215, 87.0 | 247 | - | ||||
| DM treatment (n, %) | 191, 88.8 | 215 | - | ||||
| Oral DM medication (n, %) | 180, 83.7 | 215 | - | ||||
| Insulin treatment, (n, %) | 20, 9.3 | 215 | - | ||||
| BUN (mg/dL) | 15.26 | 3.94 | 887 | 16.43 | 5.89 | 247 | * |
| Creatinine (mg/dL) | 0.93 | 0.15 | 887 | 0.99 | 0.38 | 247 | * |
| AST(SGOT) (IU/L) | 25.28 | 9.19 | 887 | 26.47 | 11.88 | 247 | n.s. |
| ALT(SGPT) (IU/L) | 22.16 | 12.60 | 886 | 27.90 | 16.19 | 247 | *** |
| Total Cholesterol (mg/dL) | 191.87 | 32.92 | 887 | 188.01 | 36.08 | 247 | n.s. |
| HDL-Cholesterol (mg/dL) | 44.68 | 11.50 | 887 | 39.51 | 8.56 | 247 | *** |
| Triglyceride (mg/dL) | 123.48 | 73.37 | 887 | 181.91 | 112.83 | 247 | *** |
| hs-CRP (mg/L) | 1.33 | 3.81 | 887 | 2.12 | 4.96 | 247 | n.s. |
| W.B.C. blood (Thous/uL) | 5.22 | 1.35 | 887 | 6.13 | 1.59 | 247 | *** |
| R.B.C. blood (Mil/uL) | 4.43 | 0.42 | 887 | 4.46 | 0.46 | 247 | n.s. |
| Hemoglobin (Hb) (g/dL) | 13.70 | 1.38 | 887 | 13.73 | 1.49 | 247 | n.s. |
| Hematocrit (Hct) (%) | 41.17 | 3.84 | 887 | 41.06 | 4.23 | 247 | n.s. |
| Platelet (Thous/uL) | 255.22 | 60.11 | 887 | 255.35 | 66.94 | 247 | n.s. |
| Probe | Delta-Beta | p-Value | Adj. p-Value | CHR | MAPINFO | Gene | Feature | cgi | Methylation |
|---|---|---|---|---|---|---|---|---|---|
| cg19693031 | −0.058 | 2.95 × 10−49 | 2.13 × 10−43 | 1 | 145441552 | TXNIP | 3′UTR | opensea | hypo |
| cg26974062 | −0.022 | 2.76 × 10−36 | 9.97 × 10−31 | 1 | 145440734 | TXNIP | Body | opensea | hypo |
| cg26823705 | −0.029 | 3.22 × 10−23 | 5.82 × 10−18 | 1 | 145435523 | NBPF20 | Body | opensea | hypo |
| cg04816311 | 0.021 | 2.34 × 10−20 | 2.42 × 10−15 | 7 | 1066650 | C7orf50 | Body | shore | hyper |
| cg17075888 | −0.033 | 2.52 × 10−19 | 2.03 × 10−14 | 7 | 95225339 | PDK4 | Body | shore | hypo |
| cg16740586 | 0.026 | 3.05 × 10−18 | 2.21 × 10−13 | 21 | 43655919 | ABCG1 | Body | shore | hyper |
| cg02841972 | −0.021 | 1.90 × 10−15 | 1.06 × 10−10 | 2 | 10176151 | IGR | opensea | hypo | |
| cg19750657 | 0.025 | 4.10 × 10−15 | 1.98 × 10−10 | 13 | 38935967 | UFM1 | 3′UTR | opensea | hyper |
| cg10217853 | −0.037 | 3.99 × 10−14 | 1.70 × 10−9 | 15 | 98505199 | ARRDC4 | Body | shore | hypo |
| cg00683922 | 0.021 | 4.66 × 10−14 | 1.87 × 10−9 | 1 | 207242569 | PFKFB2 | Body | opensea | hyper |
| Chromosome | Start | End | Width | p Value | p Value Area | Gene | |
|---|---|---|---|---|---|---|---|
| DMR_1 | chr6 | 32063114 | 32065211 | 2097 | 0 | 0.00040626 | TNXB |
| DMR_2 | chr6 | 30038910 | 30039600 | 690 | 0.0002208 | 0.00106864 | RNF39 |
| DMR_3 | chr5 | 135415693 | 135416613 | 920 | 0.0006271 | 0.002040131 | MIR886 |
| DMR_4 | chr10 | 530635 | 531584 | 949 | 0.0011923 | 0.003170594 | DIP2C |
| DMR_5 | chr1 | 153599479 | 153600156 | 677 | 0.0018193 | 0.004265729 | S100A13 |
| DMR_6 | chr6 | 29648161 | 29649024 | 863 | 0.002049 | 0.010306638 | |
| DMR_7 | chr6 | 33047944 | 33048879 | 935 | 0.0022698 | 0.005581658 | HLA-DPB1 |
| DMR_8 | chr6 | 31691354 | 31692152 | 798 | 0.0024464 | 0.017168898 | C6orf25 |
| DMR_9 | chr6 | 31275551 | 31275881 | 330 | 0.0026937 | 0.006924082 | |
| DMR_10 | chr16 | 875257 | 875626 | 369 | 0.0026672 | 0.037163952 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seo, H.; Park, J.-H.; Hwang, J.-T.; Choi, H.-K.; Park, S.-H.; Lee, J. Epigenetic Profiling of Type 2 Diabetes Mellitus: An Epigenome-Wide Association Study of DNA Methylation in the Korean Genome and Epidemiology Study. Genes 2023, 14, 2207. https://doi.org/10.3390/genes14122207
Seo H, Park J-H, Hwang J-T, Choi H-K, Park S-H, Lee J. Epigenetic Profiling of Type 2 Diabetes Mellitus: An Epigenome-Wide Association Study of DNA Methylation in the Korean Genome and Epidemiology Study. Genes. 2023; 14(12):2207. https://doi.org/10.3390/genes14122207
Chicago/Turabian StyleSeo, Hyein, Jae-Ho Park, Jin-Taek Hwang, Hyo-Kyoung Choi, Soo-Hyun Park, and Jangho Lee. 2023. "Epigenetic Profiling of Type 2 Diabetes Mellitus: An Epigenome-Wide Association Study of DNA Methylation in the Korean Genome and Epidemiology Study" Genes 14, no. 12: 2207. https://doi.org/10.3390/genes14122207