
The Complete Mitochondrial Genome of Torix tukubana (Annelida: Hirudinea: Glossiphoniidae)


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Genome Assembly and Annotation
2.3. Phylogenetic Analysis
3. Results and Discussion
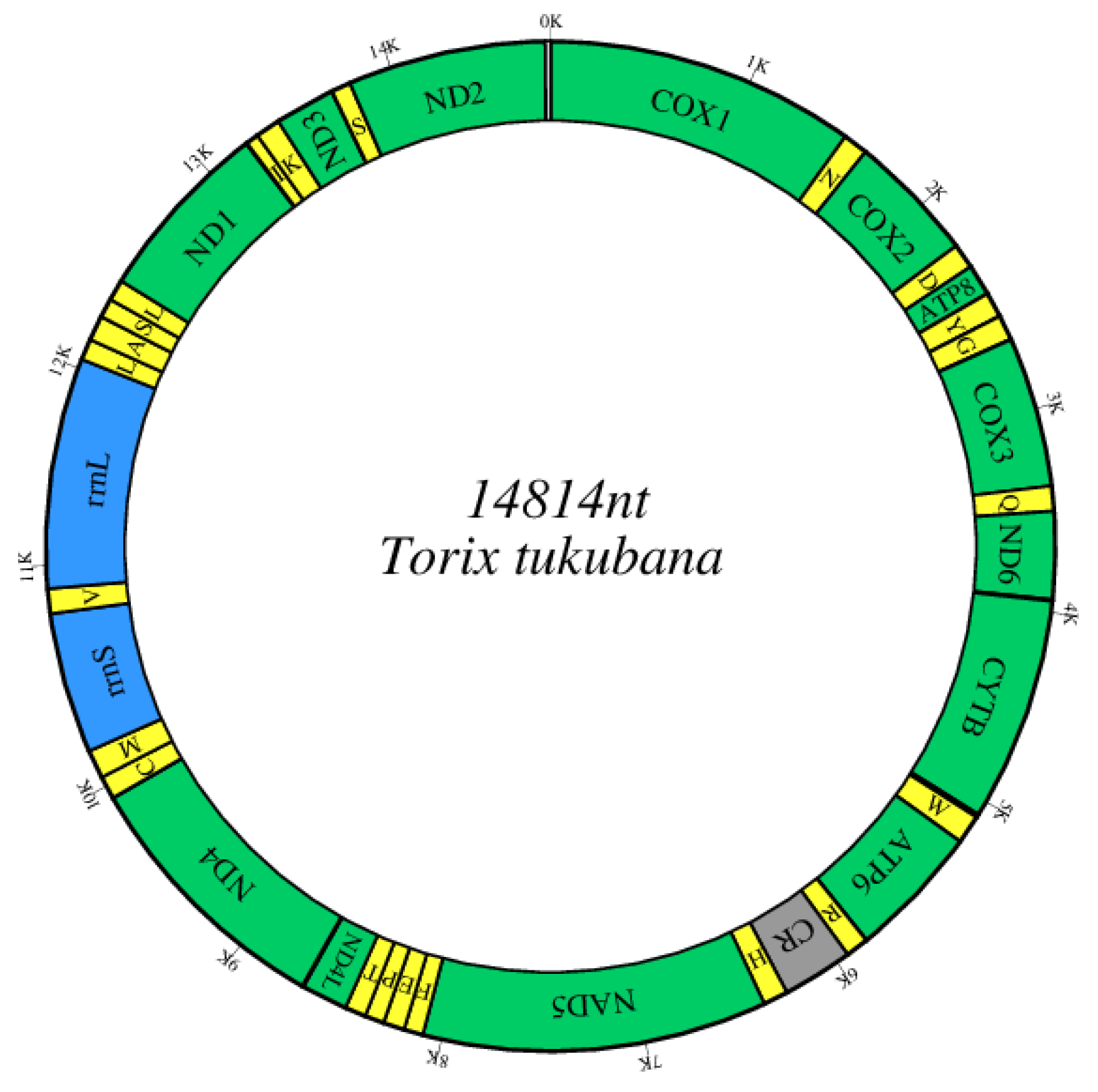
3.1. Genome Composition
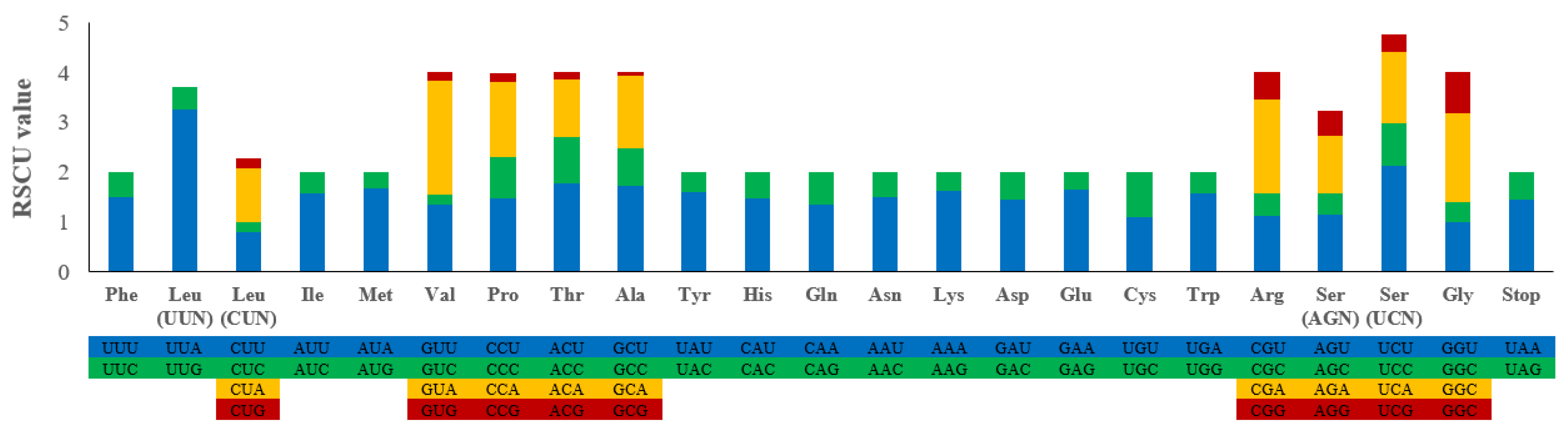
3.2. Protein-Coding Genes
3.3. tRNAs, rRNAs, and CR
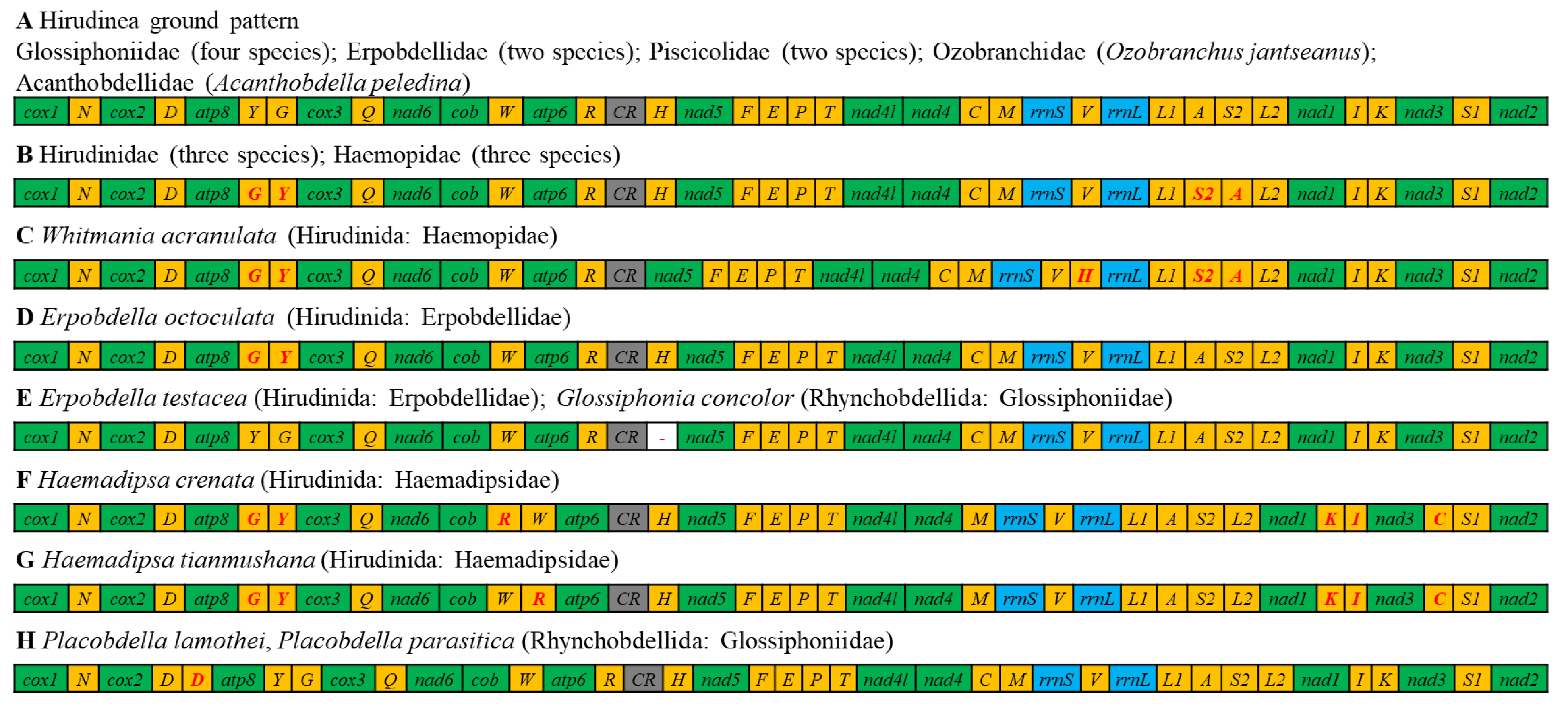
3.4. Gene Arrangement
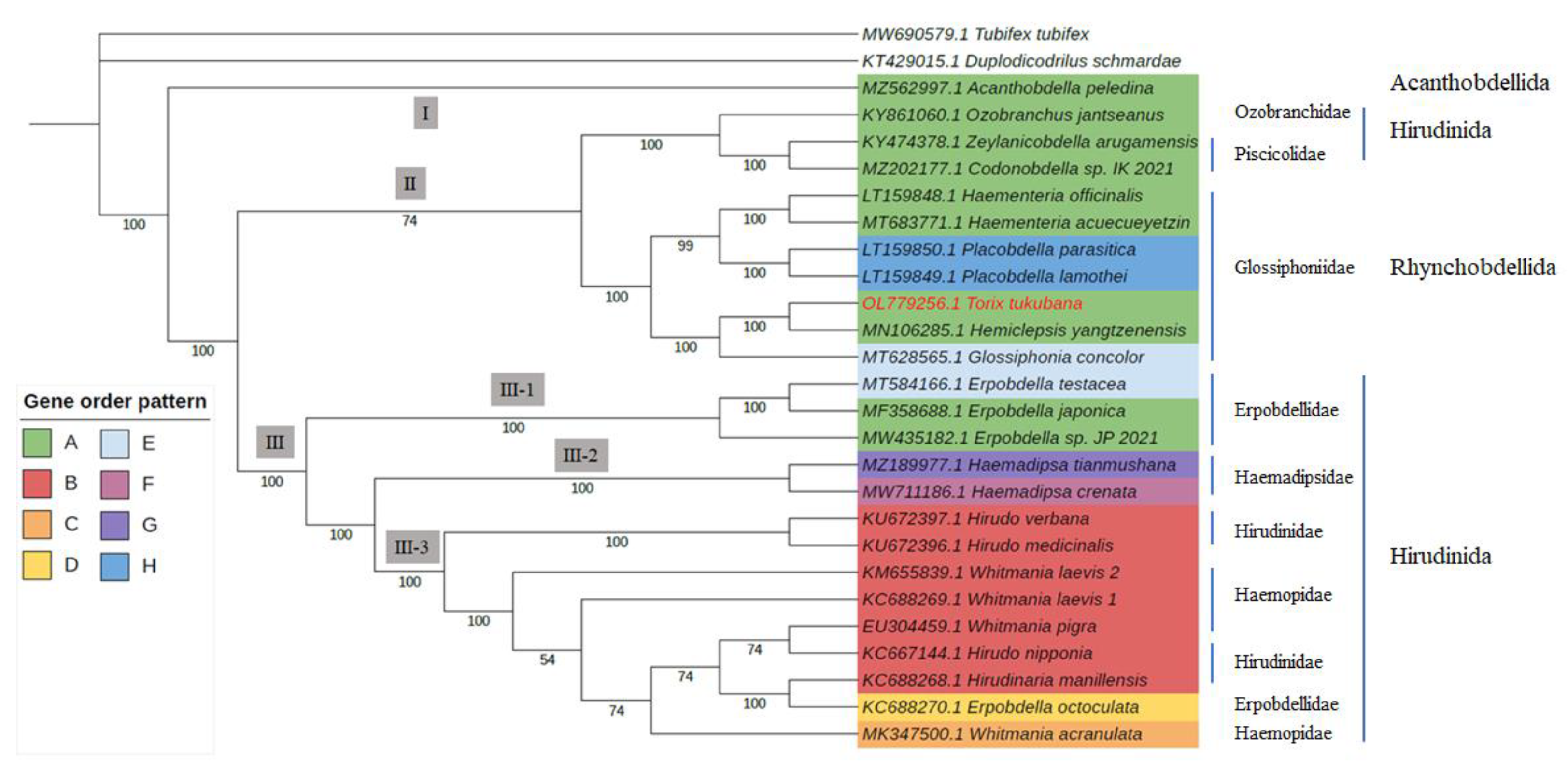
3.5. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Apakupakul, K.; Siddall, M.E.; Burreson, E.M. Higher Level Relationships of Leeches (Annelida: Clitellata: Euhirudinea) Based on Morphology and Gene Sequences. Mol. Phylogenet. Evol. 1999, 12, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Moser, W.E.; Govedich, F.R.; Klemm, D.J. Annelida, Hirudinida (Leeches). In Encyclopedia of Inland Waters; Elsevier: Amsterdam, The Netherlands, 2009; pp. 116–123. ISBN 978-0-12-370626-3. [Google Scholar]
- Oka, A. Notices Sur Les Hirudinées d’Extreme Orient. Anno. Zool. Jpn 1925, 10, 311–326. [Google Scholar]
- Simon, C.; Frati, F.; Beckenbach, A.; Crespi, B.; Liu, H.; Flook, P. Evolution, Weighting, and Phylogenetic Utility of Mitochondrial Gene Sequences and a Compilation of Conserved Polymerase Chain Reaction Primers. Ann. Entomol. Soc. Am. 1994, 87, 651–701. [Google Scholar] [CrossRef]
- Boore, J.L. Animal Mitochondrial Genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Shen, X.; Wu, Z.; Sun, M.; Ren, J.; Liu, B. The Complete Mitochondrial Genome Sequence of Whitmania pigra (Annelida, Hirudinea): The First Representative from the Class Hirudinea. Comp. Biochem. Physiol. Part D Genom. Proteom. 2011, 6, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Dabney, J.; Knapp, M.; Glocke, I.; Gansauge, M.-T.; Weihmann, A.; Nickel, B.; Valdiosera, C.; García, N.; Pääbo, S.; Arsuaga, J.L.; et al. Complete Mitochondrial Genome Sequence of a Middle Pleistocene Cave Bear Reconstructed from Ultrashort DNA Fragments. Proc. Natl. Acad. Sci. USA 2013, 110, 15758–15763. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhu, X.; Li, Y.; Han, Z.; Xu, W.; Dong, J.; Wei, H.; Li, X. Mitochondrial Genome of Chinese Grass Shrimp, Palaemonetes sinensis and Comparison with Other Palaemoninae Species. Sci. Rep. 2019, 9, 17301. [Google Scholar] [CrossRef] [PubMed]
- Roe, A.D.; Sperling, F.A.H. Patterns of Evolution of Mitochondrial Cytochrome c Oxidase I and II DNA and Implications for DNA Barcoding. Mol. Phylogenet. Evol. 2007, 44, 325–345. [Google Scholar] [CrossRef]
- Boore, J.L.; Brown, W.M. Big Trees from Little Genomes: Mitochondrial Gene Order as a Phylogenetic Tool. Curr. Opin. Genet. Dev. 1998, 8, 668–674. [Google Scholar] [CrossRef]
- Boore, J.L.; Macey, J.R.; Medina, M. Sequencing and Comparing Whole Mitochondrial Genomes of Animals. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2005; Volume 395, pp. 311–348. ISBN 978-0-12-182800-4. [Google Scholar]
- Boore, J.L.; Lavrov, D.V.; Brown, W.M. Gene Translocation Links Insects and Crustaceans. Nature 1998, 392, 667–668. [Google Scholar] [CrossRef]
- Sankoff, D.; Leduc, G.; Antoine, N.; Paquin, B.; Lang, B.F.; Cedergren, R. Gene Order Comparisons for Phylogenetic Inference: Evolution of the Mitochondrial Genome. Proc. Natl. Acad. Sci. USA 1992, 89, 6575–6579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Feng, H.; Tu, W.; Xiong, C.; Jin, X.; Li, P.; Wang, X.; Li, Q. Comparative Mitogenomic Analysis Reveals Dynamics of Intron Within and Between Tricholoma Species and Phylogeny of Basidiomycota. Front. Genet. 2021, 12, 534871. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Liu, T.; Zhu, W.; You, P. Complete Mitochondrial Genome of Whitmania laevis (Annelida, Hirudinea) and Comparative Analyses within Whitmania Mitochondrial Genomes. Belg. J. Zool. 2015, 145, 114–128. [Google Scholar] [CrossRef]
- Liu, X.; Luo, D.; Zhao, Y.; Zhang, Q.; Zhang, J. Complete Mithochondrial Genome of Ozobranchus jantseanus (Hirudinida: Arhychobdellida: Ozobranchidae). Mitochondrial DNA Part B 2017, 2, 232–233. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Tong, X.; Su, Y.; Meng, F.; Liu, Z. Characterization of the Complete Mitogenome of a Land Leech, Haemadipsa crenata Ngamprasertwong (Arhynchobdellida: Haemadipsidae). Mitochondrial DNA Part B 2021, 6, 2069–2070. [Google Scholar] [CrossRef]
- Kambayashi, C.; Kurabayashi, A.; Nakano, T. Topotype-Based Redescription of the Leech Torix tukubana (Hirudinida: Glossiphoniiformes: Glossiphoniidae). Proc. Biol. Soc. Wash. 2020, 133, 59. [Google Scholar] [CrossRef]
- Kambayashi, C.; Kurabayashi, A.; Nakano, T. Evaluating the Ontogenetic External Morphology of an Ectoparasitic Torix tukubana (Hirudinida: Glossiphoniidae), with Records of Its New Host Amphibian Species. Parasitol. Res. 2019, 118, 663–666. [Google Scholar] [CrossRef]
- Kikuchi, Y.; Fukatsu, T. Rickettsia Infection in Natural Leech Populations. Microb. Ecol. 2005, 49, 265–271. [Google Scholar] [CrossRef]
- Siddall, M.E.; Burreson, E.M. Phylogeny of Leeches (Hirudinea) Based on Mitochondrial Cytochromec Oxidase Subunit I. Mol. Phylogenet. Evol. 1998, 9, 156–162. [Google Scholar] [CrossRef]
- Borda, E.; Siddall, M.E. Insights into the Evolutionary History of Indo-Pacific Blood feeding Terrestrial Leeches (Hirudinida: Arhynchobdellida: Haemadipisdae). Invert. Syst. 2010, 24, 456. [Google Scholar] [CrossRef]
- Schnell, I.B.; Bohmann, K.; Schultze, S.E.; Richter, S.R.; Murray, D.C.; Sinding, M.-H.S.; Bass, D.; Cadle, J.E.; Campbell, M.J.; Dolch, R.; et al. Debugging Diversity—A Pan-Continental Exploration of the Potential of Terrestrial Blood-Feeding Leeches as a Vertebrate Monitoring Tool. Mol. Ecol. Resour. 2018, 18, 1282–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utevsky, S.; Kovalchuk, A.; Kovalchuk, N.; Utevsky, A.; Chernyshev, A.V. A New Species of the Genus Johanssonia selensky, 1914 (Hirudinea: Piscicolidae) Collected in the Kuril-Kamchatka Trench at the Greatest Depth Ever Recorded for Fish Leeches. Prog. Oceanogr. 2019, 176, 102133. [Google Scholar] [CrossRef]
- Schubert, M.; Lindgreen, S.; Orlando, L. AdapterRemoval v2: Rapid Adapter Trimming, Identification, and Read Merging. BMC Res. Notes 2016, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An Empirically Improved Memory-Efficient Short-Read de Novo Assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Coil, D.; Jospin, G.; Darling, A.E. A5-Miseq: An Updated Pipeline to Assemble Microbial Genomes from Illumina MiSeq Data. Bioinformatics 2015, 31, 587–589. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and Open Software for Comparing Large Genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and Accurate Annotation of Organelle Genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de Novo Metazoan Mitochondrial Genome Annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perna, N.T.; Kocher, T.D. Patterns of Nucleotide Composition at Fourfold Degenerate Sites of Animal Mitochondrial Genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A Fast Online Phylogenetic Tool for Maximum Likelihood Analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (ITOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Oceguera-Figueroa, A.; Manzano-Marín, A.; Kvist, S.; Moya, A.; Siddall, M.E.; Latorre, A. Comparative Mitogenomics of Leeches (Annelida: Clitellata): Genome Conservation and Placobdella-Specific TrnD Gene Duplication. PLoS ONE 2016, 11, e0155441. [Google Scholar] [CrossRef]
- Xu, Z.; Yang, C.; Gofarov, M.Y.; Eliseeva, T.A.; Kondakov, A.V.; Yuan, H.; Bolotov, I.N.; Yang, D. A New Freshwater Leech Species from Asian Swamp Eel Stocks in China. Parasitol. Res. 2021, 120, 2769–2778. [Google Scholar] [CrossRef]
- Borda, E.; Siddall, M.E. Arhynchobdellida (Annelida: Oligochaeta: Hirudinida): Phylogenetic Relationships and Evolution. Mol. Phylogenet. Evol. 2004, 30, 213–225. [Google Scholar] [CrossRef]
- Nikitina, A.; Babenko, V.; Akopian, T.; Shirokov, D.; Manuvera, V.; Kurdyumov, A.; Kostryukova, E.; Lazarev, V. Draft Mitochondrial Genomes of Hirudo medicinalis and Hirudo verbana (Annelida, Hirudinea). Mitochondrial DNA Part B 2016, 1, 254–256. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Accession No. | Size (bp) | Nucleotide Composition/% | AT Skew | GC Skew | ||||
|---|---|---|---|---|---|---|---|---|---|
| T(U) | C | A | G | A + T (U) | |||||
| T. tukubana | OL779256 | 14,814 | 38.4 | 14.5 | 35.2 | 11.9 | 73.6 | −0.043 | −0.098 |
| Acanthobdella peledina | MZ562997.1 | 18,528 | 35.2 | 19.0 | 34.7 | 11.1 | 69.9 | −0.007 | −0.262 |
| Codonobdella sp. IK-2021 | MZ202177.1 | 14,486 | 38.9 | 12.3 | 36.6 | 12.2 | 75.5 | −0.030 | −0.004 |
| Erpobdella japonica | MF358688.1 | 14,725 | 37.0 | 14.9 | 35.2 | 13.0 | 72.2 | −0.025 | −0.068 |
| Erpobdella octoculata | KC688270.1 | 14,407 | 40.6 | 12.5 | 30.9 | 15.9 | 71.5 | −0.136 | 0.120 |
| Erpobdella sp. JP-2021 | MW435182.1 | 15,469 | 36.0 | 17.4 | 33.8 | 12.8 | 69.8 | −0.032 | −0.152 |
| Erpobdella testacea * | MT584166.1 | 14,495 | 36.7 | 14.5 | 36.4 | 12.4 | 73.1 | −0.004 | −0.078 |
| Glossiphonia concolor * | MT628565.1 | 14,548 | 39.2 | 13.1 | 35.5 | 12.2 | 74.7 | −0.050 | −0.036 |
| Haemadipsa crenata | MW711186.1 | 14,725 | 42.4 | 10.9 | 34.4 | 12.3 | 76.8 | −0.104 | 0.060 |
| Haemadipsa tianmushana | MZ189977.1 | 14,625 | 42.8 | 10.5 | 35.1 | 11.6 | 77.9 | −0.099 | 0.050 |
| Haementeria acuecueyetzin | MT683771.1 | 14,985 | 38.9 | 14.9 | 34.8 | 11.4 | 73.7 | −0.056 | −0.133 |
| Haementeria officinalis * | LT159848.1 | 14,849 | 39.0 | 15.1 | 34.3 | 11.7 | 73.3 | −0.064 | −0.127 |
| Hemiclepsis yangtzenensis | MN106285.1 | 14,984 | 37.7 | 15.0 | 35.2 | 12.1 | 72.9 | −0.034 | −0.107 |
| Hirudinaria manillensis | KC688268.1 | 14,470 | 40.7 | 12.2 | 31.4 | 15.8 | 72.1 | −0.129 | 0.129 |
| Hirudo medicinalis * | KU672396.1 | 14,729 | 42.8 | 10.7 | 33.4 | 13.2 | 76.2 | −0.123 | 0.105 |
| Hirudo nipponia | KC667144.1 | 14,414 | 40.9 | 11.9 | 31.7 | 15.5 | 72.6 | −0.127 | 0.131 |
| Hirudo verbana * | KU672397.1 | 14,604 | 43.2 | 10.5 | 33.7 | 12.7 | 76.9 | −0.124 | 0.095 |
| Ozobranchus jantseanus | KY861060.1 | 14,864 | 37.4 | 14.9 | 35.0 | 12.6 | 72.4 | −0.033 | −0.084 |
| Placobdella lamothei | LT159849.1 | 15,190 | 36.4 | 18.0 | 31.7 | 13.9 | 68.1 | −0.069 | −0.129 |
| Placobdella parasitica * | LT159850.1 | 14,909 | 37.8 | 15.5 | 34 | 12.7 | 71.8 | −0.053 | −0.099 |
| Whitmania acranulata | MK347500.1 | 14,468 | 40.8 | 12.1 | 30.8 | 16.3 | 71.6 | −0.140 | 0.148 |
| Whitmania laevis 1 | KC688269.1 | 14,433 | 41.2 | 12.0 | 30.8 | 160. | 72.0 | −0.144 | 0.143 |
| Whitmania laevis 2 | KM655839.1 | 14,442 | 41.9 | 11.1 | 31.1 | 15.9 | 73.0 | −0.148 | 0.178 |
| Whitmania pigra | EU304459.1 | 14,426 | 41.3 | 11.8 | 30.8 | 16.1 | 72.1 | −0.146 | 0.154 |
| Zeylanicobdella arugamensis | KY474378.1 | 16,161 | 43.7 | 10.4 | 35.5 | 10.3 | 79.2 | −0.104 | −0.005 |
| Gene | Position | Gene Length/bp | Start Codon | Stop Codon | Anti Codon | H/L Strand | Intergenic/Overlapping Region Length/bp |
|---|---|---|---|---|---|---|---|
| cox1 | 1–1537 | 1537 | ATG | T-- | + | ||
| trnN | 1538–1601 | 64 | GTT | + | |||
| cox2 | 1602–2279 | 678 | ATG | TAA | + | 1 | |
| trnD | 2281–2344 | 64 | GTC | + | |||
| atp8 | 2345–2503 | 159 | ATG | TAG | + | −2 | |
| trnY | 2502–2567 | 66 | GTA | + | −2 | ||
| trnG | 2566–2626 | 61 | TCC | + | −3 | ||
| cox3 | 2624–3404 | 781 | ATA | T-- | + | ||
| trnQ | 3405–3473 | 69 | TTG | + | 1 | ||
| nad6 | 3475–3945 | 471 | ATG | TAA | + | −8 | |
| cob | 3938–5074 | 1137 | ATG | TAA | + | 16 | |
| trnW | 5091–5154 | 64 | TCA | + | 1 | ||
| atp6 | 5156–5860 | 705 | ATG | TAA | + | −2 | |
| trnR | 5859–5926 | 68 | TCG | + | |||
| CR | 5927–6328 | 402 | |||||
| trnH | 6329–6392 | 64 | GTG | + | |||
| nad5 | 6393–8094 | 1702 | ATG | T-- | + | ||
| trnF | 8095–8157 | 63 | GAA | + | |||
| trnE | 8158–8216 | 59 | TTC | + | 1 | ||
| trnP | 8218–8281 | 64 | TGG | + | |||
| trnT | 8282–8343 | 62 | TGT | + | |||
| nad4l | 8344–8637 | 294 | ATG | TAA | + | −7 | |
| nad4 | 8631–9965 | 1335 | ATG | TAG | + | −2 | |
| trnC | 9964–10,027 | 64 | GCA | + | |||
| trnM | 10,028–10,089 | 62 | CAT | + | |||
| rrnS | 10,090–10,824 | 735 | + | ||||
| trnV | 10,825–10,887 | 63 | TAC | + | |||
| rrnL | 10,888–12,047 | 1160 | + | ||||
| trnL1 | 12,048–12,107 | 60 | TAG | + | |||
| trnA | 12,108–12,167 | 60 | TGC | + | −1 | ||
| trnS2 | 12,167–12,233 | 67 | TGA | + | |||
| trnL2 | 12,234–12,294 | 61 | TAA | + | |||
| nad1 | 12,295–13,233 | 939 | ATG | TAA | + | 5 | |
| trnI | 13,239–13,300 | 62 | GAT | + | |||
| trnK | 13,301–13,365 | 65 | TTT | + | |||
| nad3 | 13,366–13,719 | 354 | ATG | TAG | + | −3 | |
| trnS1 | 13,717–13,782 | 66 | TCT | + | 1 | ||
| nad2 | 13,784–14,788 | 1005 | ATT | TAA | + | 25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, X.; Zhao, Y.; Wei, H.; Hu, N.; Hu, Q.; Li, Y. The Complete Mitochondrial Genome of Torix tukubana (Annelida: Hirudinea: Glossiphoniidae). Genes 2023, 14, 388. https://doi.org/10.3390/genes14020388
Zhu X, Zhao Y, Wei H, Hu N, Hu Q, Li Y. The Complete Mitochondrial Genome of Torix tukubana (Annelida: Hirudinea: Glossiphoniidae). Genes. 2023; 14(2):388. https://doi.org/10.3390/genes14020388
Chicago/Turabian StyleZhu, Xiaochen, Yingying Zhao, Hua Wei, Nan Hu, Qingbiao Hu, and Yingdong Li. 2023. "The Complete Mitochondrial Genome of Torix tukubana (Annelida: Hirudinea: Glossiphoniidae)" Genes 14, no. 2: 388. https://doi.org/10.3390/genes14020388