
ThrRS-Mediated Translation Regulation of the RNA Polymerase III Subunit RPC10 Occurs through an Element with Similarity to Cognate tRNA ASL and Affects tRNA Levels
Abstract
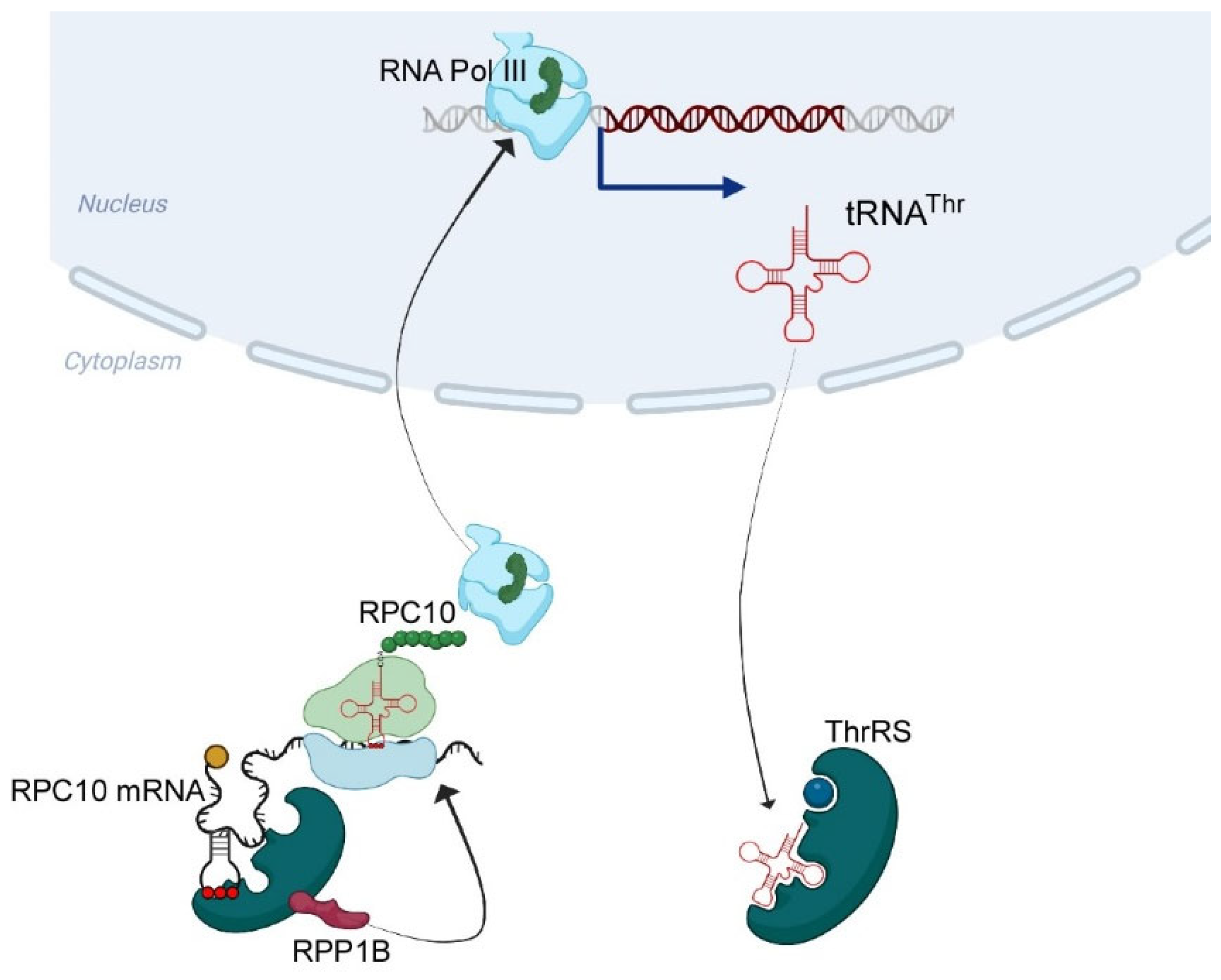
:1. Introduction
2. Results
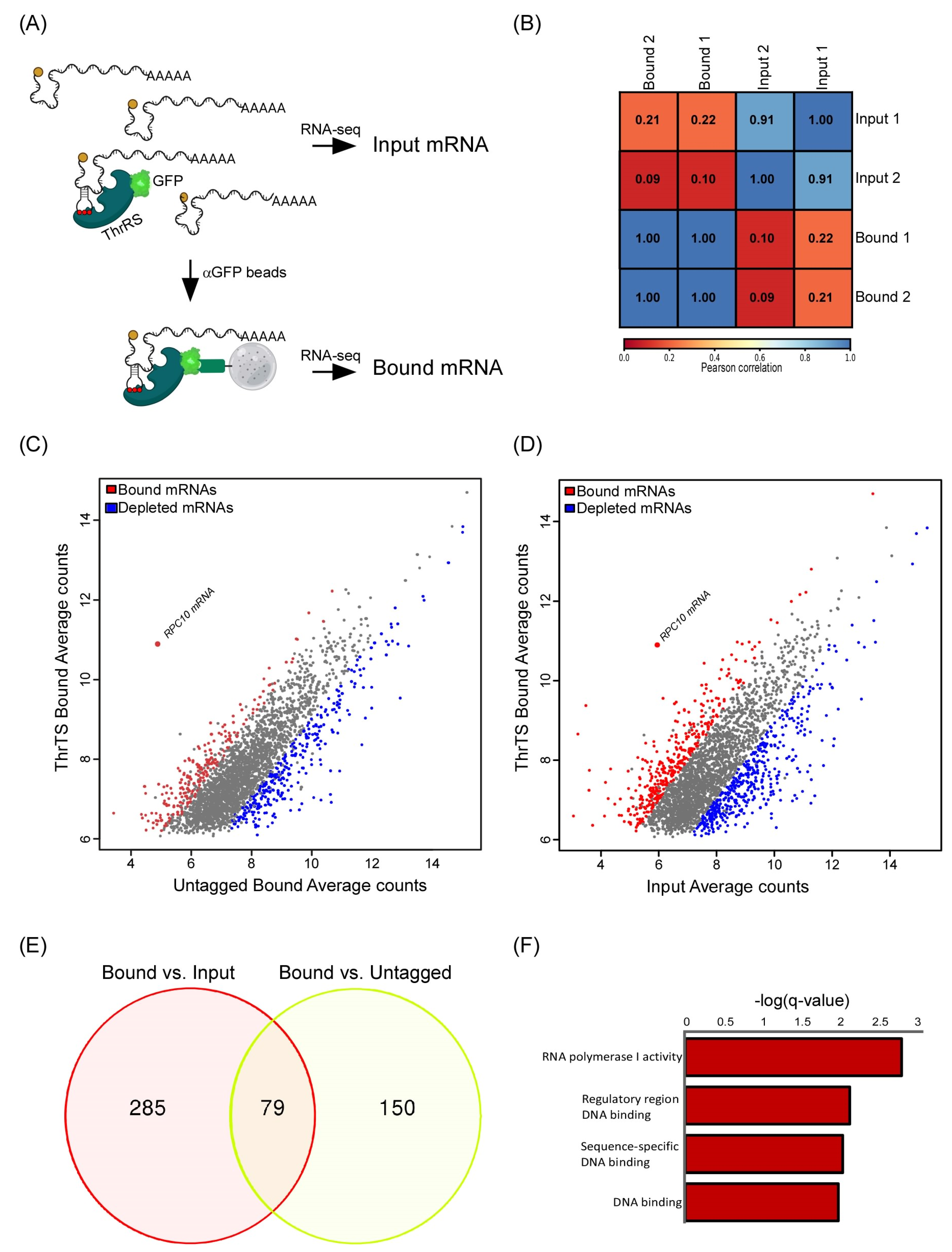
2.1. mRNAs Bound by ThrRS
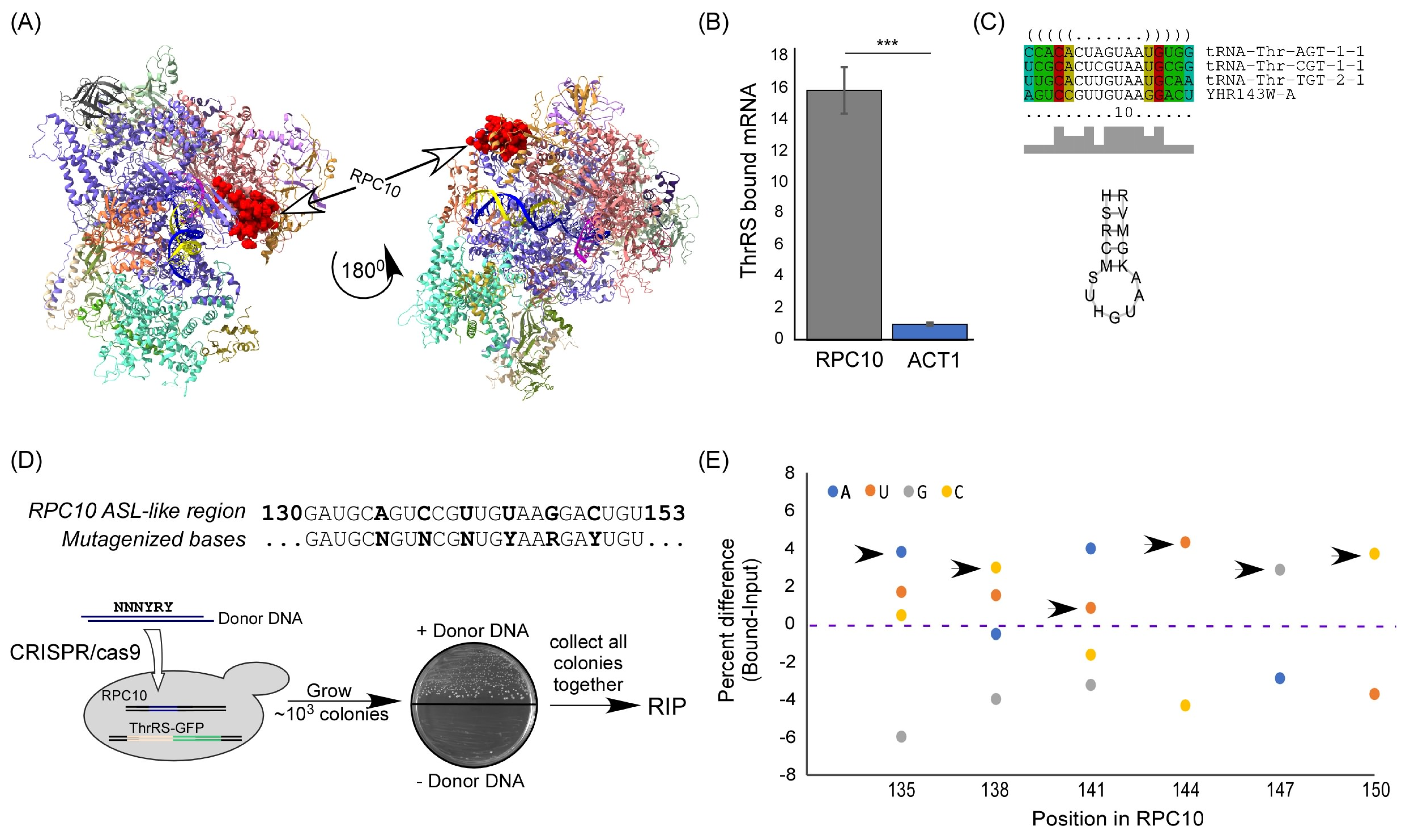
2.2. RPC10 Is Bound by ThrRS through an Element Predicted to Form Cognate tRNA ASL
2.3. Altered ThrRS Binding upon Elimination of the ASL-like Element
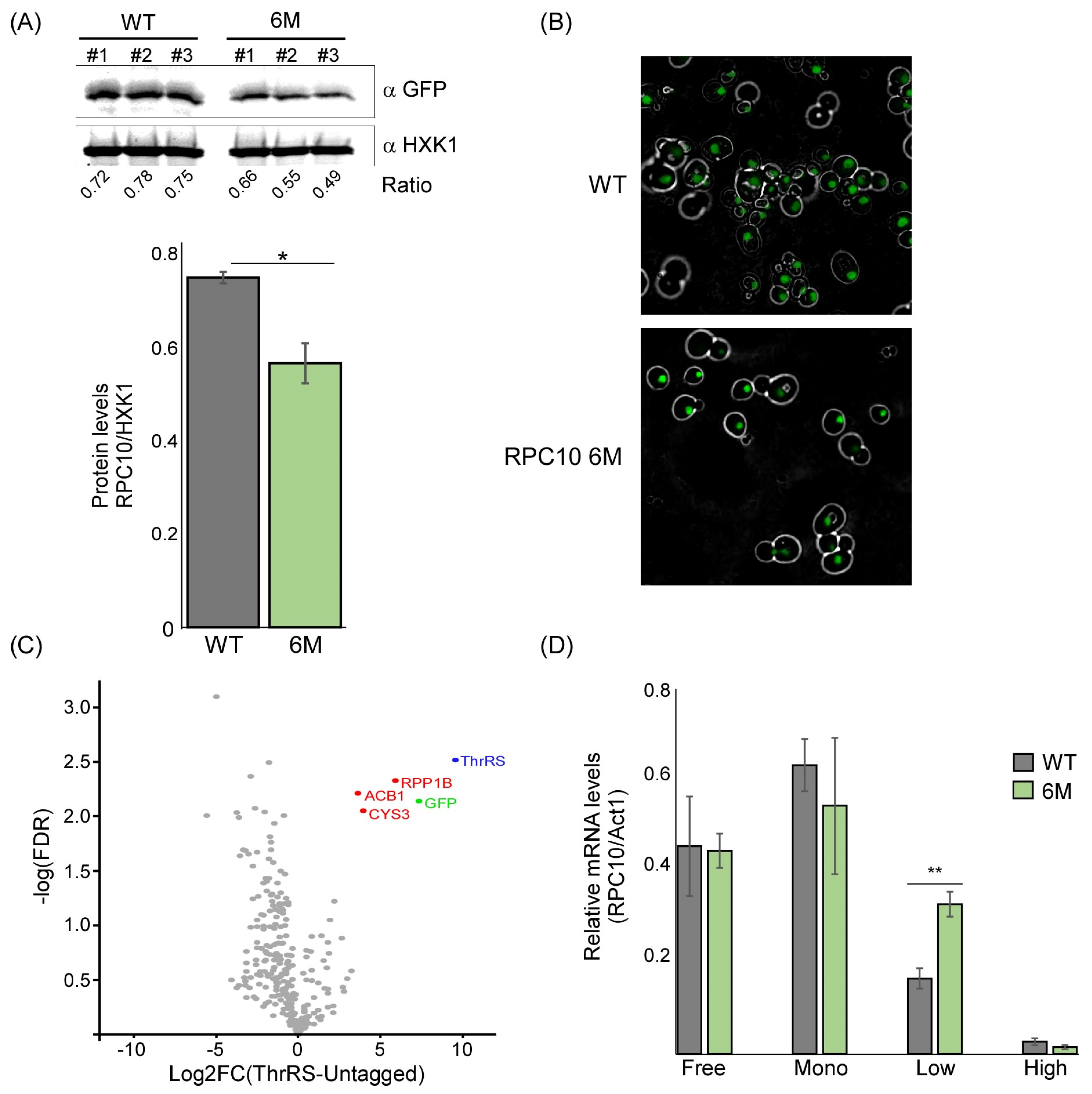
2.4. The Predicted ASL-Mimic Is Important for RPC10 Translation
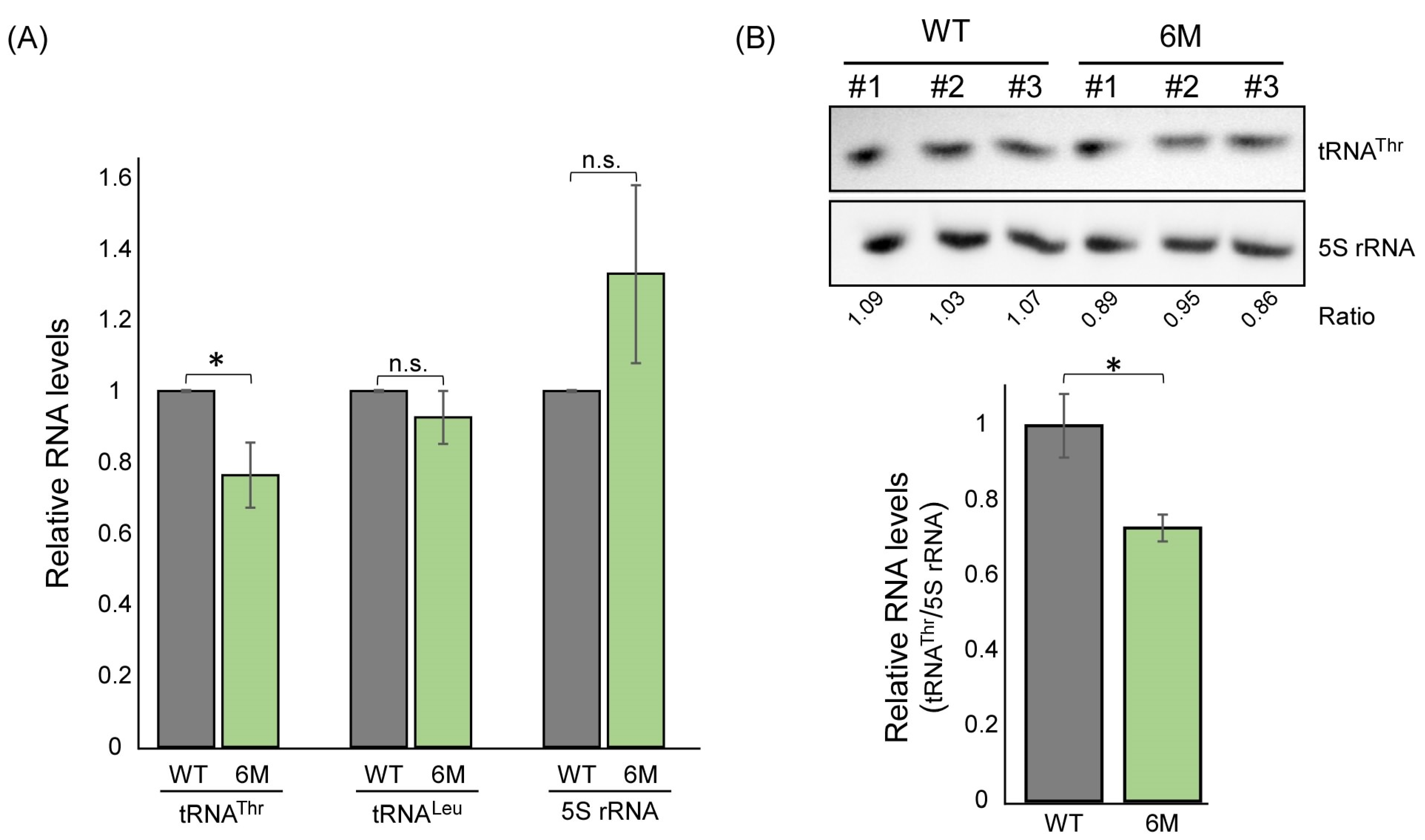
2.5. Mutations within the Predicted ASL-like Site Lead to an Impact on tRNAThr Expression
3. Discussion
3.1. The Importance of the Predicted ASL-like for Translation Regulation of RPC10
3.2. Regulation of Expression by tRNA Mimicry
3.3. Regulation of tRNA Expression by ThrRS
4. Materials and Methods
4.1. Yeast Strains, Growth Conditions, and Plasmids
4.2. RBP ImmunoPrecipitation (RIP) and RNA-seq Analyses
4.3. CRISPR/Cas9-Mediated Recombination
4.4. Polysome Analysis
4.5. tRNA Quantification by RT-qPCR
4.6. Protein Co-Immunoprecipitation
4.7. Mass Spectrometry (MS/MS) Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lab Code | Strain Name | Description | Genotype | Source |
|---|---|---|---|---|
| YA1 | BY4741 | Wild type | Mat a, his3∆1, leu2∆0, met15∆0,ura3∆0 | [39] |
| YA1545 | THS1-GFP | THS1 (YIL078W) C-terminally tagged with GFP, in BY4741 | MATa, leu2Δ0, met15Δ0, ura3Δ0, THS1: GFP (S65T)-HIS3 | [40] |
| YA1595 | THS1-GFP RPC10-6M | THS1 (YIL078W) C-terminally tagged with GFP, BY4741. RPC10 gene was mutated (AGTCCGTTGTAAGGAC to GGTGCGGTGCAAAGAT) | MATa, leu2Δ0, met15Δ0, ura3Δ0, THS1: GFP (S65T)-HIS3, rpc10-6m | This study |
| YA1619 | GFP-RPC10 | N terminally GFP-tagged RPC10 (YHR143W-A), BY4741 | MATa, leu2Δ0, met15Δ0, ura3Δ0, GFP(S65T)-HIS3: RPC10 | [41] |
| YA1620 | GFP-RPC10 6M | N terminally GFP-tagged RPC10 (YHR143W-A), BY4741. RPC10 gene was mutated (AGTCCGTTGTAAGGAC to GGTGCGGTGCAAAGAT) | MATa, leu2Δ0, met15Δ0, ura3Δ0, GFP(S65T)-HIS3: RPC10 | This study |
| Lab Code | Plasmid Name | Description | Source |
|---|---|---|---|
| pA1067 | bRA66 | (Empty Backbone) GAL1 driven Cas9 Addgene #100952 | [42] |
| pA1194 | RPC10 + 143 gRNA | bRA66 with RPC10 +143 (AACTGATGCAGTCCGTTGTA) targeting gRNA cloned into BplI sites | This study |
| gRNA Oligos | |
| Primer Name | Primer sequence |
| RPC10 gRNA-143 F | AACTGATGCAGTCCGTTGTAGTTTT |
| RPC10 gRNA-143 R | TACAACGGACTGCATCAGTTGATCA |
| Donor DNA Oligos | |
| Primer Name | Primer sequence |
| RPC10 Random Donor DNA F | TAGTAAATTATCTTTATCCAGAACTGATGCNGTNCGNTGYAARGAYTGTGGTCATAGAATCCTGTTGAAGGCTAGGACTA |
| RPC10 Random Donor DNA R | TAGTCCTAGCCTTCAACAGGATTCTATGACCACARTCYTTRCANCGNACNGCATCAGTTCTGGATAAAGATAATTTACTA |
| RPC10 6M Donor DNA F | TAGTAAATTATCTTTATCCAGAACTGATGCgGTgCGgTGcAAaGAtTGTGGTCATAGAATCCTGTTGAAGGCTAGGACTA |
| RPC10 6M Donor DNA R | TAGTCCTAGCCTTCAACAGGATTCTATGACCACAaTCtTTgCAcCGcACcGCATCAGTTCTGGATAAAGATAATTTACTA |
| qPCR Oligos | |
| Primer Name | Primer sequence |
| RPC10 seq F | ATGTCTCGCGAAGGGTTC |
| RPC10 seq R | TCAAATTGAACCAATCTCTTAGTCC |
| RT-qPCR Oligos | |
| Primer Name | Primer sequence |
| RPC10 F | CGCGAAGGGTTCCAGATT |
| RPC10 R | CTTCAAAGTTGCCGTTCTAGC |
| Act1 F | GATTCTGAGGTTGCTGCTTTG |
| Act1 R | ACCGACGATAGATGGGAAGA |
| tRNA-Thr-AGT F | CAAGTTGGTAAGGCGCCAC |
| tRNA-Thr-AGT R | TCGGATTTGAACCGATGATCTCC |
| tRNA-Leu-CAA F | TTTGGCCGAGCGGTCTAAGG |
| tRNA-Leu-CAA R | TGCATCTTACGATACCTGAGCTT |
| 5S rRNA F | GCGGCCATATCTACCAGAAA |
| 5S rRNA R | GTATGGTCACCCACTACACTAC |
| Northern probes | |
| tRNAThr biotin | /5BiosG/GGATTTGAACCGATGATCT |
| 5S biotin | /5BiosG/TGGTAGATATGGCCGCAACC |
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gomez, M.A.R.; Ibba, M. Aminoacyl-tRNA synthetases. RNA 2020, 26, 910–936. [Google Scholar] [CrossRef] [PubMed]
- Giegé, R.; Sissler, M.; Florentz, C. Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res. 1998, 26, 5017–5035. [Google Scholar] [CrossRef] [PubMed]
- Giegé, R.; Eriani, G. Transfer RNA Recognition and Aminoacylation by Synthetases; Wiley: New York, NY, USA, 2014. [Google Scholar] [CrossRef]
- Guo, M.; Schimmel, P. Essential nontranslational functions of tRNA synthetases. Nat. Chem. Biol. 2013, 9, 145–153. [Google Scholar] [CrossRef]
- Pang, Y.L.J.; Poruri, K.; Martinis, S.A. tRNA synthetase: tRNA aminoacylation and beyond. Wiley Interdiscip. Rev. RNA 2014, 5, 461–480. [Google Scholar] [CrossRef] [PubMed]
- Levi, O.; Garin, S.; Arava, Y. RNA mimicry in post-transcriptional regulation by aminoacyl tRNA synthetases. WIREs RNA 2019, 11, e1564. [Google Scholar] [CrossRef]
- Sampath, P.; Mazumder, B.; Seshadri, V.; Gerber, C.A.; Chavatte, L.; Kinter, M.; Ting, S.M.; Dignam, J.D.; Kim, S.; Driscoll, D.M.; et al. Noncanonical Function of Glutamyl-Prolyl-tRNA Synthetase. Cell 2004, 119, 195–208. [Google Scholar] [CrossRef]
- Garin, S.; Levi, O.; Forrest, M.E.; Antonellis, A.; Arava, Y.S. Comprehensive characterization of mRNAs associated with yeast cytosolic aminoacyl-tRNA synthetases. RNA Biol. 2021, 18, 2605–2616. [Google Scholar] [CrossRef]
- Ryckelynck, M.; Giegé, R.; Frugier, M. tRNAs and tRNA mimics as cornerstones of aminoacyl-tRNA synthetase regulations. Biochimie 2005, 87, 835–845. [Google Scholar] [CrossRef]
- Moine, H.; Romby, P.; Springer, M.; Grunberg-Manago, M.; Ebel, J.P.; Ehresmann, B.; Ehresmann, C. Escherichia coli threonyl-tRNA synthetase and tRNA(Thr) modulate the binding of the ribosome to the translational initiation site of the thrS mRNA. J. Mol. Biol. 1990, 216, 299–310. [Google Scholar] [CrossRef]
- Levi, O.; Arava, Y. mRNA association by aminoacyl tRNA synthetase occurs at a putative anticodon mimic and autoregulates translation in response to tRNA levels. PLoS Biol. 2019, 17, e3000274. [Google Scholar] [CrossRef]
- Treich, I.; Carles, C.; Riva, M.; Sentenac, A. RPC10 encodes a new mini subunit shared by yeast nuclear RNA polymerases. Gene Expr. 1992, 2, 31–37. [Google Scholar] [PubMed]
- Huang, Y.; Maraia, R.J. Comparison of the RNA polymerase III transcription machinery in Schizosaccharomyces pombe, Saccharomyces cerevisiae and human. Nucleic Acids Res. 2001, 29, 2675–2690. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, S.; Wan, F.; Xu, Y.; Wu, Z.; Cao, M.; Lan, P.; Lei, M.; Wu, J. Structural insights into transcriptional regulation of human RNA polymerase III. Nat. Struct. Mol. Biol. 2021, 28, 220–227. [Google Scholar] [CrossRef]
- Rubbi, L.; Labarre-Mariotte, S.; Chédin, S.; Thuriaux, P. Functional Characterization of ABC10α, an Essential Polypeptide Shared by All Three Forms of Eukaryotic DNA-dependent RNA Polymerases. J. Biol. Chem. 1999, 274, 31485–31492. [Google Scholar] [CrossRef]
- Will, S.; Joshi, T.; Hofacker, I.L.; Stadler, P.F.; Backofen, R. LocARNA-P: Accurate boundary prediction and improved detection of structural RNAs. RNA 2012, 18, 900–914. [Google Scholar] [CrossRef]
- Levi, O.; Arava, Y. Expanding the CRISPR/Cas9 Toolbox for Gene Engineering in S. cerevisiae. Curr. Microbiol. 2020, 77, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Zuk, D.; Belk, J.P.; Jacobson, A. Temperature-sensitive mutations in the Saccharomyces cerevisiae MRT4, GRC5, SLA2 and THS1 genes result in defects in mRNA turnover. Genetics 1999, 153, 35–47. [Google Scholar] [CrossRef]
- Sabi, R.; Daniel, R.V.; Tuller, T. stAIcalc: tRNA adaptation index calculator based on species-specific weights. Bioinformatics 2017, 33, 589–591. [Google Scholar] [CrossRef]
- Ballesta, J.P.; Remacha, M. The large ribosomal subunit stalk as a regulatory element of the eukaryotic translational machinery. Prog. Nucleic Acid Res. Mol. Biol. 1996, 55, 157–193. [Google Scholar] [CrossRef]
- Gonzalo, P.; Reboud, J.P. The puzzling lateral flexible stalk of the ribosome. Biol. Cell 2003, 95, 179–193. [Google Scholar] [CrossRef]
- Arava, Y.; Wang, Y.; Storey, J.D.; Liu, C.L.; Brown, P.O.; Herschlag, D. Genome-wide analysis of mRNA translation profiles in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2003, 100, 3889–3894. [Google Scholar] [CrossRef] [PubMed]
- Nameki, N. Identity elements of tRNA(Thr) towards Saccharomyces cerevisiae threonyl-tRNA synthetase. Nucleic Acids Res. 1995, 23, 2831–2836. [Google Scholar] [CrossRef]
- Romby, P.; Caillet, J.; Ebel, C.; Sacerdot, C.; Graffe, M.; Eyermann, F.; Brunel, C.; Moine, H.; Ehresmann, C.; Ehresmann, B.; et al. The expression of E.coli threonyl-tRNA synthetase is regulated at the translational level by symmetrical operator-repressor interactions. EMBO J. 1996, 15, 5976–5987. [Google Scholar] [CrossRef]
- Torres-Larios, A.; Dock-Bregeon, A.C.; Romby, P.; Rees, B.; Sankaranarayanan, R.; Caillet, J.; Springer, M.; Ehresmann, C.; Ehresmann, B.; Moras, D. Structural basis of translational control by Escherichia coli threonyl tRNA synthetase. Nat. Struct. Biol. 2002, 9, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.J.; Park, S.; Nguyen, L.T.; Hwang, J.; Lee, E.Y.; Giong, H.K.; Lee, J.S.; Yoon, I.; Lee, J.H.; Kim, J.H.; et al. A threonyl-tRNA synthetase-mediated translation initiation machinery. Nat. Commun. 2019, 10, 1357. [Google Scholar] [CrossRef] [PubMed]
- Nusspaumer, G.; Remacha, M.; Ballesta, J.P.G. Phosphorylation and N-terminal region of yeast ribosomal protein P1 mediate its degradation, which is prevented by protein P2. EMBO J. 2000, 19, 6075–6084. [Google Scholar] [CrossRef]
- Remacha, M.; Jimenez-Diaz, A.; Santos, C.; Briones, E.; Zambrano, R.; Rodriguez Gabriel, M.A.; Guarinos, E.; Ballesta, J.P. Proteins P1, P2, and P0, components of the eukaryotic ribosome stalk. New structural and functional aspects. Biochem. Cell Biol. 1995, 73, 959–968. [Google Scholar] [CrossRef]
- Ames, B.N.; Tsang, T.H.; Buck, M.; Christman, M.F. The leader mRNA of the histidine attenuator region resembles tRNAHis: Possible general regulatory implications. Proc. Natl. Acad. Sci. USA 1983, 80, 5240–5242. [Google Scholar] [CrossRef]
- Brunel, C.; Romby, P.; Moine, H.; Caillet, J.; Grunberg-Manago, M.; Springer, M.; Ehresmann, B.; Ehresmann, C. Translational regulation of the Escherichia coli threonyl-tRNA synthetase gene: Structural and functional importance of the thrS operator domains. Biochimie 1993, 75, 1167–1179. [Google Scholar] [CrossRef]
- Frugier, M.; Giegé, R. Yeast aspartyl-tRNA synthetase binds specifically its own mRNA. J. Mol. Biol. 2003, 331, 375–383. [Google Scholar] [CrossRef]
- Ryckelynck, M.; Masquida, B.; Giegé, R.; Frugier, M. An intricate RNA structure with two tRNA-derived motifs directs complex formation between yeast aspartyl-tRNA synthetase and its mRNA. J. Mol. Biol. 2005, 354, 614–629. [Google Scholar] [CrossRef] [PubMed]
- Nomura, M.; Gourse, R.; Baughman, G. Regulation of the synthesis of ribosomes and ribosomal components. Annu. Rev. Biochem. 1984, 53, 75–117. [Google Scholar] [CrossRef] [PubMed]
- Nomura, M.; Yates, J.L.; Dean, D.; Post, L.E. Feedback regulation of ribosomal protein gene expression in Escherichia coli: Structural homology of ribosomal RNA and ribosomal protein MRNA. Proc. Natl. Acad. Sci. USA 1980, 77, 7084–7088. [Google Scholar] [CrossRef]
- Boguta, M. Assembly of RNA polymerase III complex involves a putative co-translational mechanism. Gene 2022, 824, 146394. [Google Scholar] [CrossRef] [PubMed]
- Dumay, H.; Rubbi, L.; Sentenac, A.; Marck, C. Interaction between yeast RNA polymerase III and transcription factor TFIIIC via ABC10alpha and tau131 subunits. J. Biol. Chem. 1999, 274, 33462–33468. [Google Scholar] [CrossRef]
- Melamed, D.; Eliyahu, E.; Arava, Y. Exploring translation regulation by global analysis of ribosomal association. Methods 2009, 48, 301–305. [Google Scholar] [CrossRef]
- Torrent, M.; Chalancon, G.; De Groot, N.S.; Wuster, A.; Madan Babu, M. Cells alter their tRNA abundance to selectively regulate protein synthesis during stress conditions. Sci. Signal. 2018, 11, eaat6409. [Google Scholar] [CrossRef]
- Winston, F.; Dollard, C.; Ricupero-Hovasse, S.L. Construction of a set of convenient Saccharomyces cerevisiae strains that are isogenic to S288C. Yeast 1995, 11, 53–55. [Google Scholar] [CrossRef]
- Huh, W.K.; Falvo, J.V.; Gerke, L.C.; Carroll, A.S.; Howson, R.W.; Weissman, J.S.; O’Shea, E.K. Global analysis of protein localization in budding yeast. Nature 2003, 425, 686–691. [Google Scholar] [CrossRef]
- Weill, U.; Yofe, I.; Sass, E.; Stynen, B.; Davidi, D.; Natarajan, J.; Ben-Menachem, R.; Avihou, Z.; Goldman, O.; Harpaz, N.; et al. Genome-wide SWAp-Tag yeast libraries for proteome exploration. Nat. Methods 2018, 15, 617–622. [Google Scholar] [CrossRef]
- Anand, R.; Beach, A.; Li, K.; Haber, J. Rad51-mediated double-strand break repair and mismatch correction of divergent substrates. Nature 2017, 544, 377–380. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levi, O.; Mallik, M.; Arava, Y.S. ThrRS-Mediated Translation Regulation of the RNA Polymerase III Subunit RPC10 Occurs through an Element with Similarity to Cognate tRNA ASL and Affects tRNA Levels. Genes 2023, 14, 462. https://doi.org/10.3390/genes14020462
Levi O, Mallik M, Arava YS. ThrRS-Mediated Translation Regulation of the RNA Polymerase III Subunit RPC10 Occurs through an Element with Similarity to Cognate tRNA ASL and Affects tRNA Levels. Genes. 2023; 14(2):462. https://doi.org/10.3390/genes14020462
Chicago/Turabian StyleLevi, Ofri, Monalisha Mallik, and Yoav S. Arava. 2023. "ThrRS-Mediated Translation Regulation of the RNA Polymerase III Subunit RPC10 Occurs through an Element with Similarity to Cognate tRNA ASL and Affects tRNA Levels" Genes 14, no. 2: 462. https://doi.org/10.3390/genes14020462