
PeCLH2 Gene Positively Regulate Salt Tolerance in Transgenic Populus alba × Populus glandulosa


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Genetic Phylogenetic Analysis
2.3. Molecular Cloning of the Gene
2.4. GUS Experiment
2.5. Breeding of Transgenic Plants with Overexpression and RNA Interference Expression
2.6. Morphometry
2.7. Transgenic Salt Stress Experiment, Physiological Index Determination and Histochemical Staining
2.8. Expression Analysis of Stress Resistance Related Genes
2.9. RNA Sequencing
2.10. GO and KEGG Enrichment Analyses of DEGs
2.11. RT-qPCR Examination of RNA Sequencing Results
3. Result
3.1. Phylogenetic Analysis and Gene Cloning
3.2. Expression Analysis of PeCLH2
3.3. Gus Staining
3.4. Identification of Transgenic Plants
3.5. Morphological Characteristics of Transgenic Plants
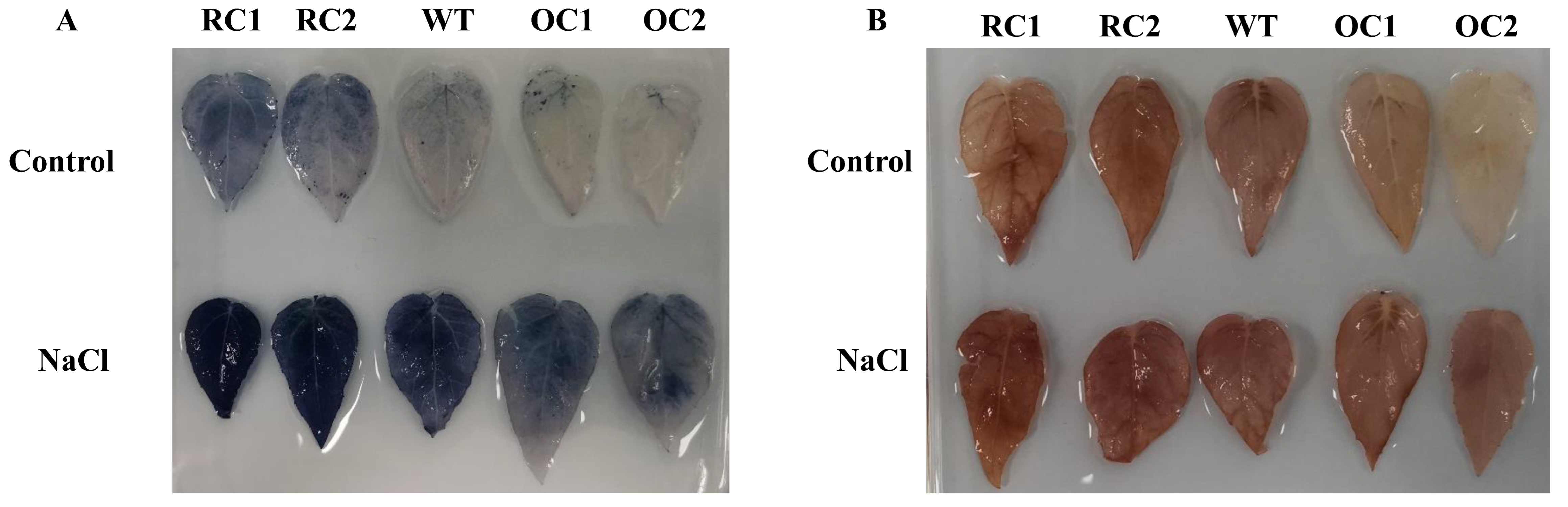
3.6. Physiological Index Determination and Histochemical Staining Analysis
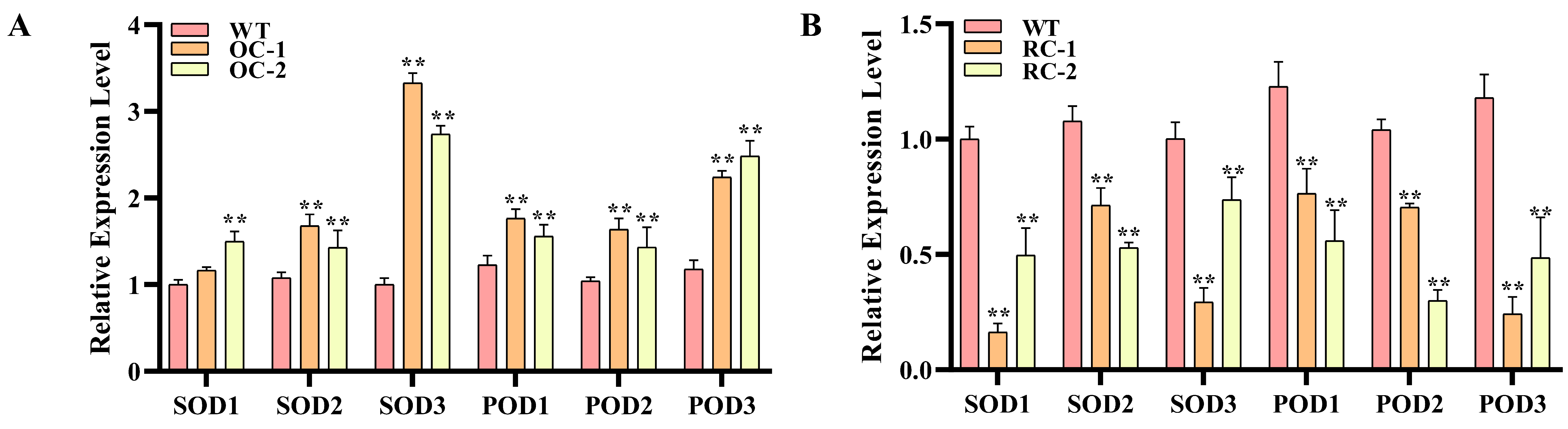
3.7. Expression Analysis of Stress Resistance Related Genes
3.8. Sequencing Data Quality Assessment
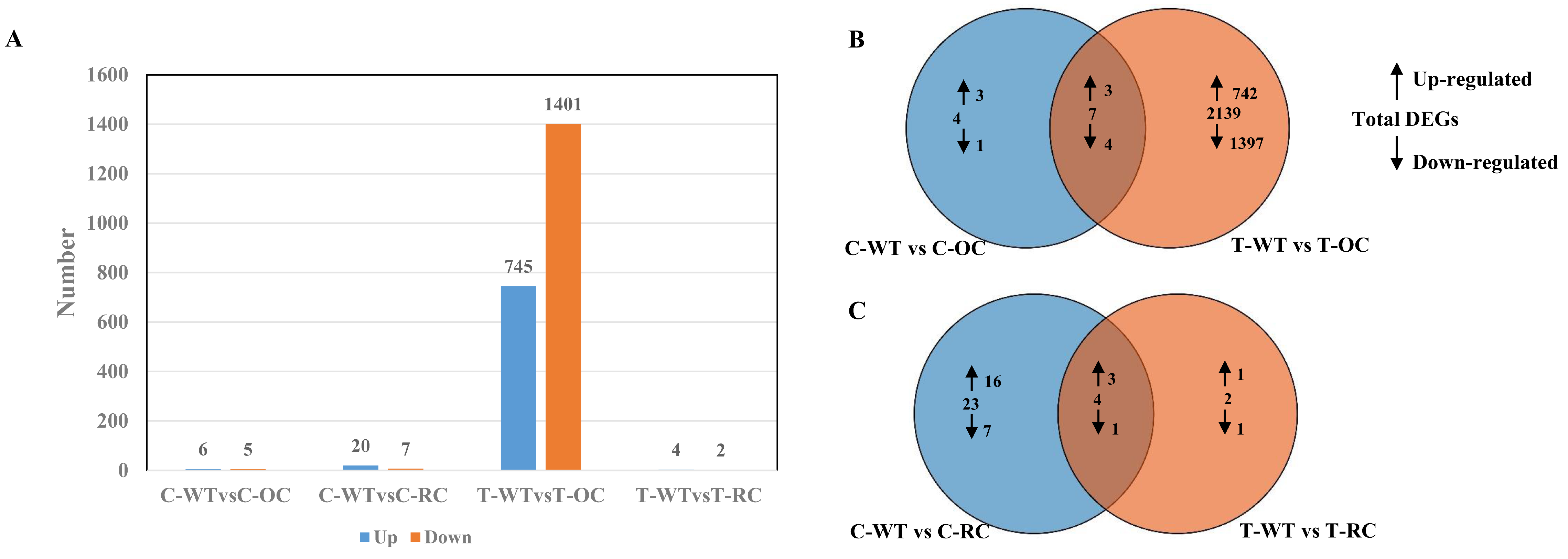
3.9. Gene Expression Level Analysis
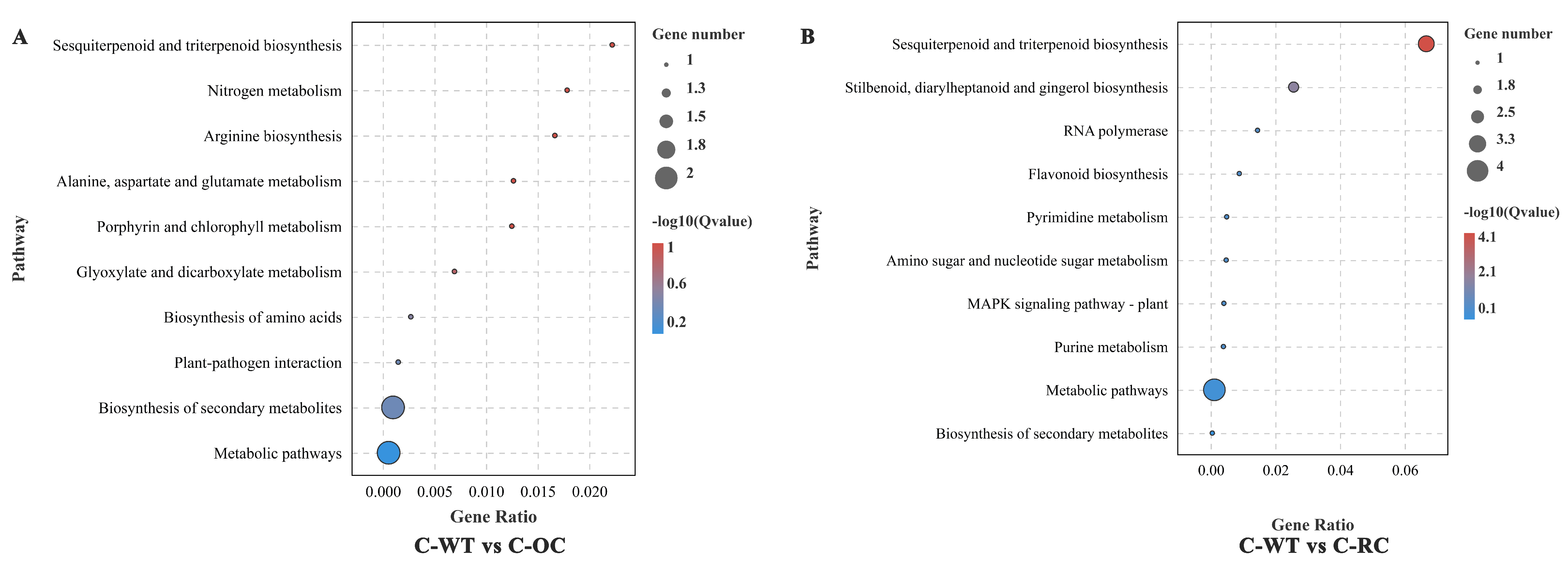
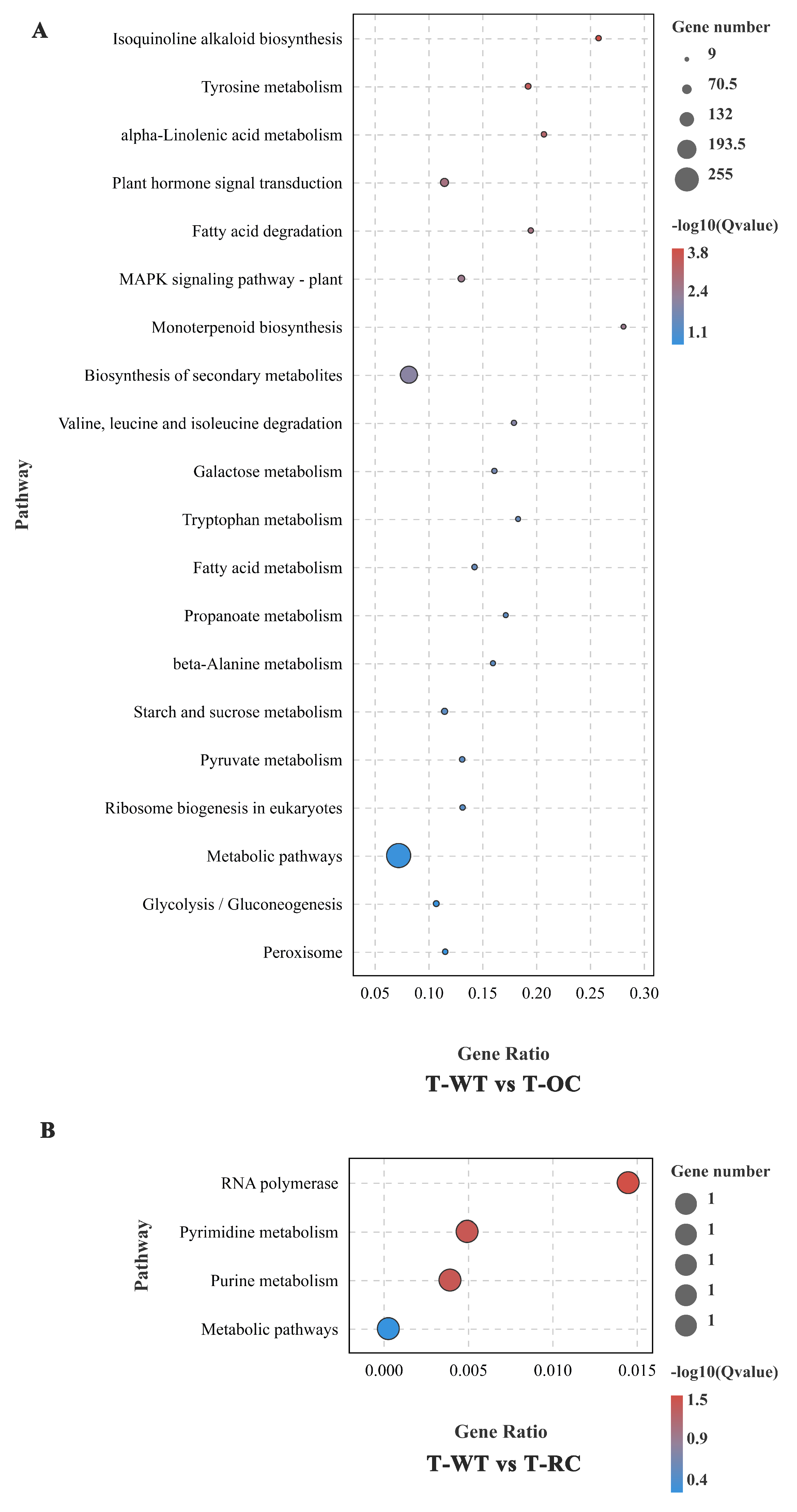
3.10. GO and KEGG Enrichment Analyses of DEGs
3.11. Transcription Factors Involved in Salt Stress Response
3.12. Reliability of Transcriptome Sequencing Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Acosta-Motos, J.; Ortuño, M.; Bernal-Vicente, A.; Diaz-Vivancos, P.; Sanchez-Blanco, M.; Hernandez, J. Plant responses to salt stress: Adaptive mechanisms. Agronomy 2017, 7, 18. [Google Scholar] [CrossRef] [Green Version]
- Sunita, K.; Mishra, I.; Mishra, J.; Prakash, J.; Arora, N. Secondary metabolites from halotolerant plant growth promoting rhizobacteria for ameliorating salinity stress in plants. Front. Microbiol. 2020, 11, 567768. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Druzhinina, I.; Pan, X.; Yuan, Z. Microbially mediated plant salt tolerance and microbiome-based solutions for saline agriculture. Biotechnol. Adv. 2016, 34, 1245–1259. [Google Scholar] [CrossRef] [PubMed]
- Dresselhaus, T.; Hückelhoven, R. Biotic and abiotic stress responses in crop plants. Agronomy 2018, 8, 267. [Google Scholar] [CrossRef] [Green Version]
- Jing, X.; Cai, C.; Fan, S.; Wang, L.; Zeng, X. Spatial and temporal calcium signaling and its physiological effects in Moso Bamboo under drought stress. Forests 2019, 10, 224. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Han, X.; Yang, T.; Cui, W.H.; Wu, A.M.; Fu, C.X.; Wang, B.C.; Liu, L. Genome-wide transcriptional adaptation to salt stress in Populus. BMC Plant Biol. 2019, 19, 367. [Google Scholar] [CrossRef]
- Rao, S.; Du, C.; Li, A.; Xia, X.; Yin, W.; Chen, J. Salicylic acid alleviated salt damage of Populus euphratica: A physiological and transcriptomic analysis. Forests 2019, 10, 423. [Google Scholar] [CrossRef] [Green Version]
- Deng, S.; Sun, J.; Zhao, R.; Ding, M.; Zhang, Y.; Sun, Y.; Wang, W.; Tan, Y.; Liu, D.; Ma, X.; et al. Populus euphratica APYRASE2 enhances cold tolerance by modulating vesicular trafficking and extracellular ATP in Arabidopsis plants. Plant Physiol. 2015, 169, 530–548. [Google Scholar] [CrossRef] [Green Version]
- Shen, C.; Zhang, Y.; Li, Q.; Liu, S.; He, F.; An, Y.; Zhou, Y.; Liu, C.; Yin, W.; Xia, X. PdGNC confers drought tolerance by mediating stomatal closure resulting from NO and H2O2 production via the direct regulation of PdHXK1 expression in Populus. New Phytol. 2021, 230, 1868–1882. [Google Scholar] [CrossRef]
- Hauenstein, M.; Christ, B.; Das, A.; Aubry, S.; Hortensteiner, S. A role for TIC55 as a hydroxylase of phyllobilins, the products of chlorophyll breakdown during plant senescence. Plant Cell 2016, 28, 2510–2527. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.; Ding, S.; Zhang, L.; Xu, D.; Zhang, Y.; Sun, M.; Ge, Y.; Li, J. Low temperature delays degreening of apple fruit by inhibiting pheophorbide a oxygenase (PAO) pathway and chlorophyll oxidation during ripening. J. Food Biochem. 2022, 46, e14173. [Google Scholar] [CrossRef] [PubMed]
- Mattila, H.; Valev, D.; Havurinne, V.; Khorobrykh, S.; Virtanen, O.; Antinluoma, M.; Mishra, K.; Tyystjarvi, E. Degradation of chlorophyll and synthesis of flavonols during autumn senescence-the story told by individual leaves. AoB Plants 2018, 10, ply028. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Gu, T.; Khan, I.; Zada, A.; Jia, T. Research progress in the interconversion, turnover and degradation of chlorophyll. Cells 2021, 10, 3134. [Google Scholar] [CrossRef] [PubMed]
- Azoulay Shemer, T.; Harpaz-Saad, S.; Belausov, E.; Lovat, N.; Krokhin, O.; Spicer, V.; Standing, E.; Goldschmidt, E.; Eyal, Y. Citrus chlorophyllase dynamics at ethylene-induced fruit color-break: A study of chlorophyllase expression, posttranslational processing kinetics, and in situ intracellular localization. Plant Physiol. 2008, 148, 108–118. [Google Scholar] [CrossRef] [Green Version]
- Schelbert, S.; Aubry, S.; Burla, B.; Agne, B.; Kessler, F.; Krupinska, K.; Hortensteiner, S. Pheophytin pheophorbide hydrolase (pheophytinase) is involved in chlorophyll breakdown during leaf senescence in Arabidopsis. Plant Cell 2009, 21, 767–785. [Google Scholar] [CrossRef] [Green Version]
- Harpaz-Saad, S.; Azoulay, T.; Arazi, T.; Ben-Yaakov, E.; Mett, A.; Shiboleth, Y.; Hortensteiner, S.; Gidoni, D.; Gal-On, A.; Goldschmidt, E.; et al. Chlorophyllase is a rate-limiting enzyme in chlorophyll catabolism and is posttranslationally regulated. Plant Cell 2007, 19, 1007–1022. [Google Scholar] [CrossRef] [Green Version]
- Saidi, M.; Jbir, R.; Ghorbel, I.; Namsi, A.; Drira, N.; Gargouri-Bouzid, R. Brittle leaf disease induces an oxidative stress and decreases the expression of manganese-related genes in date palm (Phoenix dactylifera L.). Plant Physiol. Biochem. PPB 2012, 50, 1–7. [Google Scholar] [CrossRef]
- Hu, X.; Makita, S.; Schelbert, S.; Sano, S.; Ochiai, M.; Tsuchiya, T.; Hasegawa, S.; Hortensteiner, S.; Tanaka, A.; Tanaka, R. Reexamination of chlorophyllase function implies its involvement in defense against chewing herbivores. Plant Physiol. 2015, 167, 660–670. [Google Scholar] [CrossRef] [Green Version]
- Roychoudhury, A.; Ghosh, S.; Paul, S.; Mazumdar, S.; Das, G.; Das, S. Pre-treatment of seeds with salicylic acid attenuates cadmium chloride-induced oxidative damages in the seedlings of mungbean (Vigna radiata L. Wilczek). Acta Physiol. Plant. 2015, 38, 11. [Google Scholar] [CrossRef]
- Sytykiewicz, H.; Gabryś, B.; Dancewicz, K.; Borowiak-Sobkowiak, B.; Kmieć, K.; Łukasik, I.; Sprawka, I.; Sempruch, C.; Chrzanowski, G. Aphid-stimulated transcriptional reconfigurations of chlorophyllase-2 gene in maize (Zea mays L.) seedlings. Biochem. Syst. Ecol. 2016, 68, 178–185. [Google Scholar] [CrossRef]
- Liu, W.; Li, Q.; Wang, Y.; Wu, T.; Yang, Y.; Zhang, X.; Han, Z.; Xu, X. Ethylene response factor AtERF72 negatively regulates Arabidopsis thaliana response to iron deficiency. Biochem. Biophys. Res. Commun. 2017, 491, 862–868. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wu, T.; Li, Q.; Zhang, X.; Xu, X.; Li, T.; Han, Z.; Wang, Y. An ethylene response factor (MxERF4) functions as a repressor of Fe acquisition in Malus xiaojinensis. Sci. Rep. 2018, 8, 1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, Y.; Ko, C.; Yen, C.; Chen, L.; Shaw, J. A novel recombinant chlorophyllase1 from chlamydomonas reinhardtii for the production of chlorophyllide derivatives. J. Agric. Food Chem. 2015, 63, 9496–9503. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Okazawa, A.; Itoh, Y.; Fukusaki, E.; Kobayashi, A. Expression of chlorophyllase is not induced during autumnal yellowing in Ginkgo biloba. Z. Fur Nat. C A J. Biosci. 2004, 5–6, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, T.; Ohta, H.; Okawa, K.; Iwamatsu, A.; Shimada, H.; Masuda, T.; Takamiya, K. Cloning of chlorophyllase, the key enzyme in chlorophyll degradation: Finding of a lipase motif and the induction by methyl jasmonate. Proc. Natl. Acad. Sci. USA 1999, 96, 15362–15367. [Google Scholar] [CrossRef] [Green Version]
- Li, X.W.; Zhu, Y.L.; Chen, C.Y.; Geng, Z.J.; Li, X.Y.; Ye, T.T.; Mao, X.N.; Du, F. Cloning and characterization of two chlorophyll A/B binding protein genes and analysis of their gene family in Camellia sinensis. Sci. Rep. 2020, 10, 4602. [Google Scholar] [CrossRef] [Green Version]
- Ge, X.L.; Zhang, L.; Du, J.J.; Wen, S.S.; Qu, G.Z.; Hu, J.J. Transcriptome analysis of Populus euphratica under salt treatment and PeERF1 gene enhances salt tolerance in transgenic Populus alba x Populus glandulosa. Int. J. Mol. Sci. 2022, 23, 3727. [Google Scholar] [CrossRef]
- Jia, H.; Liu, G.; Li, J.; Zhang, J.; Sun, P.; Zhao, S.; Zhou, X.; Lu, M.; Hu, J. Genome resequencing reveals demographic history and genetic architecture of seed salinity tolerance in Populus euphratica. J. Exp. Bot. 2020, 71, 4308–4320. [Google Scholar] [CrossRef]
- Thompson, J.; Higgins, D.; Gibson, T. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.Q.; Li, Z.; Wen, S.S.; Wang, J.N.; Zhao, S.T.; Lu, M.Z. WUSCHEL-related homeobox gene PagWOX11/12a responds to drought stress by enhancing root elongation and biomass growth in poplar. J. Exp. Bot. 2020, 71, 1503–1513. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Song, X.; Wei, K.; Zhao, Y.; Jiang, C.; Wang, J.; Tang, F.; Lu, M. Growth-regulating factor 15 is required for leaf size control in Populus. Tree Physiol. 2019, 39, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Shu, W.; Zhou, H.; Jiang, C.; Zhao, S.; Wang, L.; Li, Q.; Yang, Z.; Groover, A.; Lu, M.Z. The auxin receptor TIR1 homolog (PagFBL1) regulates adventitious rooting through interactions with Aux/IAA28 in Populus. Plant Biotechnol. J. 2019, 17, 338–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, S.S.; Ge, X.L.; Wang, R.; Yang, H.F.; Bai, Y.E.; Guo, Y.H.; Zhang, J.; Lu, M.Z.; Zhao, S.T.; Wang, L.Q. An efficient agrobacterium-mediated transformation method for hybrid poplar 84K (Populus alba x P. glandulosa) using calli as explants. Int. J. Mol. Sci. 2022, 23, 2216. [Google Scholar] [CrossRef]
- Love, M.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Young, M.D.; Wakefifield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Dunlap, W.; Starcevic, A.; Baranasic, D.; Diminic, J.; Zucko, J.; Gacesa, R.; van Oppen, M.; Hranueli, D.; Cullum, J.; Long, P. KEGG orthology-based annotation of the predicted proteome of Acropora digitifera: ZoophyteBase—An open access and searchable database of a coral genome. BMC Genom. 2013, 14, 509. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Zhang, H.; Kong, L.; Gao, G.; Luo, J. PlantTFDB 3.0: A portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 2014, 42, 1182–1187. [Google Scholar] [CrossRef] [Green Version]
- Sridhar, K.; Charles, A.L. In vitro antioxidant activity of Kyoho grape extracts in DPPH and ABTS assays: Estimation methods for EC(50) using advanced statistical programs. Food Chem. 2019, 275, 41–49. [Google Scholar] [CrossRef]
- Hu, X.; Khan, I.; Jiao, Q.; Zada, A.; Jia, T. Chlorophyllase, a common plant hydrolase enzyme with a long history, is still a puzzle. Genes 2021, 12, 1871. [Google Scholar] [CrossRef]
- Chen, S.; Wang, S.; Huang, M.; Lin, K.; Hua, S.; Lu, H.; Lai, Y.; Yang, C. Physiological and molecular analyses of chlorophyllase in sweet potatoes with different-colored leaves. S. Afr. J. Bot. 2018, 114, 272–279. [Google Scholar] [CrossRef]
- Matile, P.; Hortensteiner, S.; Thomas, H. Chlorophyll degradation during senescence. Annu. Rev. Plant Biol. 2006, 57, 55–77. [Google Scholar] [CrossRef]
- Ohta, H. Degradation pathway(s) of chlorophyll: What has gene cloning revealed? Trends Plant Sci. 2000, 5, 426–431. [Google Scholar] [CrossRef]
- Shi, D.; Liu, Z.; Jin, W. Biosynthesis, catabolism and related signal regulations of plant chlorophyll. Hereditas 2009, 31, 698–704. [Google Scholar] [CrossRef]
- Gholizadeh, A. DUF538 protein superfamily is predicted to be chlorophyll hydrolyzing enzymes in plants. Physiol. Mol. Biol. Plants Int. J. Funct. Plant Biol. 2016, 22, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Atanasov, K.E.; Barboza-Barquero, L.; Tiburcio, A.F.; Alcazar, R. Genome Wide Association Mapping for the Tolerance to the Polyamine Oxidase Inhibitor Guazatine in Arabidopsis thaliana. Front. Plant Sci. 2016, 7, 401. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Luo, H.; Jiang, L.; Hou, J.; Zhang, T.; Dai, L.; Yu, Z. Integrative analysis of transcriptome and metabolome reveals the possible mechanism of leaf yellowing in pak choi (Brassica rapa subsp. chinensis) with 1-methylcyclopropene treatment during storage at 20 °C—ScienceDirect. Postharvest Biol. Technol. 2020, 169, 111300. [Google Scholar] [CrossRef]
- Zhang, S.; Wu, X.; Cui, J.; Zhang, F.; Wan, X.; Liu, Q.; Zhong, Y.; Lin, T. Physiological and transcriptomic analysis of yellow leaf coloration in Populus deltoides Marsh. PLoS ONE 2019, 14, e0216879. [Google Scholar] [CrossRef] [Green Version]
- Willsttter, R.; Stoll, A. Die Wirkungen der Chlorophyllase; Springer: Berlin/Heidelberg, Germany, 1913. [Google Scholar]
- Bose, J.; Rodrigo-Moreno, A.; Shabala, S. ROS homeostasis in halophytes in the context of salinity stress tolerance. J. Exp. Bot. 2014, 65, 1241–1257. [Google Scholar] [CrossRef]
- Mzs, A.; El-Sayed, M.; Ao, A.; Mmr, C. Pumpkin seed protein hydrolysate treatment alleviates salt stress effects on Phaseolus vulgaris by elevating antioxidant capacity and recovering ion homeostasis. Sci. Hortic. 2020, 271, 109495. [Google Scholar] [CrossRef]
- Kato, N.; Dubouzet, E.; Kokabu, Y.; Yoshida, S.; Taniguchi, Y.; Dubouzet, J.; Yazaki, K.; Sato, F. Identification of a WRKY protein as a transcriptional regulator of benzylisoquinoline alkaloid biosynthesis in Coptis japonica. Plant Cell Physiol. 2007, 48, 8–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apuya, N.; Park, J.; Zhang, L.; Ahyow, M.; Davidow, P.; Van Fleet, J.; Rarang, J.; Hippley, M.; Johnson, T.; Yoo, H.; et al. Enhancement of alkaloid production in opium and California poppy by transactivation using heterologous regulatory factors. Plant Biotechnol. J. 2008, 6, 160–175. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Kokabu, Y.; Chaki, K.; Yoshimoto, T.; Ohgaki, M.; Yoshida, S.; Kato, N.; Koyama, T.; Sato, F. Isoquinoline alkaloid biosynthesis is regulated by a unique bHLH-type transcription factor in Coptis japonica. Plant Cell Physiol. 2011, 52, 1131–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, Y.; Motomura, Y.; Sato, F. CjbHLH1 homologs regulate sanguinarine biosynthesis in Eschscholzia californica cells. Plant Cell Physiol. 2015, 56, 1019–1030. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Deng, R.; Bai, Y.; Wu, H.; Li, C.; Wu, Q.; Zhao, H. Tartary buckwheat R2R3-MYB Gene FtMYB3 negatively regulates anthocyanin and proanthocyanin biosynthesis. Int. J. Mol. Sci. 2022, 23, 2775. [Google Scholar] [CrossRef]
- Schoeffler, A.J.; Ruiz, C.R.; Joubert, A.M.; Yang, X.; LiCata, V.J. Salt modulates the stability and lipid binding affinity of the adipocyte lipid-binding proteins. J. Biol. Chem. 2003, 278, 33268–33275. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.T.; Chen, Y.L.; Zhang, X.H.; Cai, M.L.; Yu, Z.C.; Peng, C.L. ANS-deficient Arabidopsis is sensitive to high light due to impaired anthocyanin photoprotection. Funct. Plant Biol. FPB 2019, 46, 756–765. [Google Scholar] [CrossRef]
- Wang, Y.; Dong, F.; Tang, M. Transcriptome analysis of Arbuscular mycorrhizal casuarina glauca in damage mitigation of roots on NaCl stress. Microorganisms 2021, 10, 15. [Google Scholar] [CrossRef]
- Macková, H.; Hronková, M.; Dobrá, J.; Turečková, V.; Novák, O.; Lubovská, Z.; Motyka, V.; Haisel, D.; Hájek, T.; Prášil, I.T.; et al. Enhanced drought and heat stress tolerance of tobacco plants with ectopically enhanced cytokinin oxidase/dehydrogenase gene expression. J. Exp. Bot. 2013, 64, 2805–2815. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ge, X.; Du, J.; Zhang, L.; Qu, G.; Hu, J. PeCLH2 Gene Positively Regulate Salt Tolerance in Transgenic Populus alba × Populus glandulosa. Genes 2023, 14, 538. https://doi.org/10.3390/genes14030538
Ge X, Du J, Zhang L, Qu G, Hu J. PeCLH2 Gene Positively Regulate Salt Tolerance in Transgenic Populus alba × Populus glandulosa. Genes. 2023; 14(3):538. https://doi.org/10.3390/genes14030538
Chicago/Turabian StyleGe, Xiaolan, Jiujun Du, Lei Zhang, Guanzheng Qu, and Jianjun Hu. 2023. "PeCLH2 Gene Positively Regulate Salt Tolerance in Transgenic Populus alba × Populus glandulosa" Genes 14, no. 3: 538. https://doi.org/10.3390/genes14030538
APA StyleGe, X., Du, J., Zhang, L., Qu, G., & Hu, J. (2023). PeCLH2 Gene Positively Regulate Salt Tolerance in Transgenic Populus alba × Populus glandulosa. Genes, 14(3), 538. https://doi.org/10.3390/genes14030538