
A Genome-Wide Association Study to Identify Novel Candidate Genes Related to Low-Nitrogen Tolerance in Cucumber (Cucumis sativus L.)

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Treatment
2.2. Phenotype Measurement and Parameter Calculation
2.3. SNP Genotype Data Acquisition
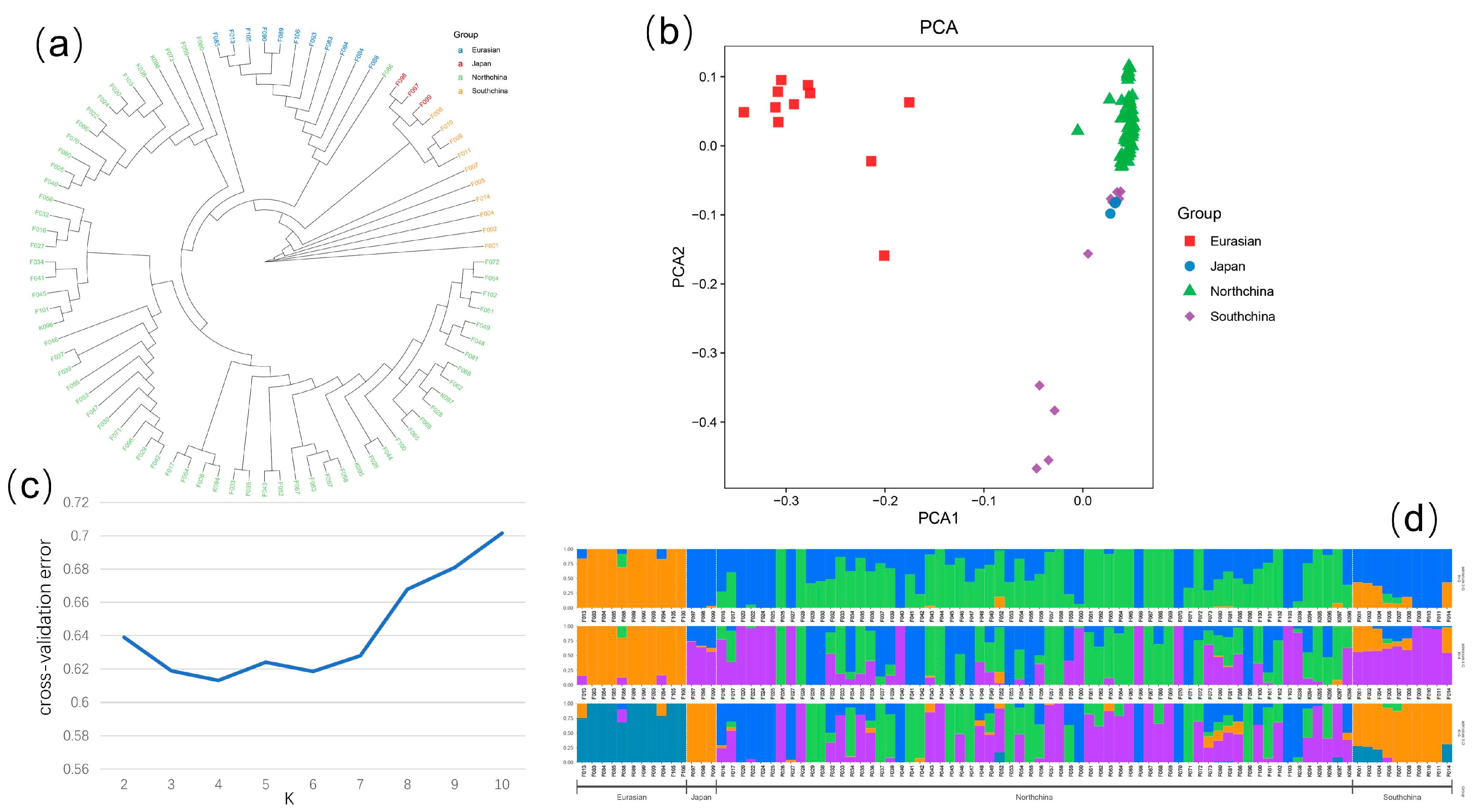
2.4. Population Genetic Evolution
2.5. GWAS
2.6. Candidate Gene Selection
3. Result
3.1. SNPs Characteristics in Cucumber Genome and Cucumber Population Distribution
3.2. Evaluation of Cucumber Population Tolerance to Low Nitrogen
3.3. GWAS
3.4. Analysis of Candidate Genes by GO Annotation
3.5. Analysis of Candidate Genes in LD blocks
3.5.1. Candidate Gene Analysis in LD block 8554
3.5.2. Candidate Gene Analysis in LD Block 1572
3.5.3. Candidate Gene Analysis in LD Block 9927
3.5.4. Candidate Gene Analysis in LD Block 2639
3.5.5. Candidate Gene Analysis in LD Block 6476
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhao, W.; Yang, X.; Yu, H.; Jiang, W.; Sun, N.; Liu, X.; Liu, X.; Zhang, X.; Wang, Y.; Gu, X. RNA-Seq-Based Transcriptome Profiling of Early Nitrogen Deficiency Response in Cucumber Seedlings Provides New Insight into the Putative Nitrogen Regulatory Network. Plant Cell Physiol. 2015, 56, 455–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Yu, H.J.; Zhang, X.M.; Yang, X.Y.; Zhao, W.C.; Li, Q.; Jiang, W.J. Effect of nitrogen deficiency on ascorbic acid biosynthesis and recycling pathway in cucumber seedlings. Plant Physiol. Biochem. 2016, 108, 222–230. [Google Scholar] [CrossRef]
- Wang, M.; Sun, Y.; Gu, Z.; Wang, R.; Sun, G.; Zhu, C.; Guo, S.; Shen, Q. Nitrate Protects Cucumber Plants Against Fusarium oxysporum by Regulating Citrate Exudation. Plant Cell Physiol. 2016, 57, 2001–2012. [Google Scholar] [CrossRef] [Green Version]
- Schulte-Uebbing, L.F.; Beusen, A.H.W.; Bouwman, A.F.; de Vries, W. From planetary to regional boundaries for agricultural nitrogen pollution. Nature 2022, 610, 507–512. [Google Scholar] [CrossRef]
- Galloway, J.N. The global nitrogen cycle: Past, present and future. Sci. China. Ser. C Life Sci. 2005, 48, 669–678. [Google Scholar]
- Liu, Q.; Wu, K.; Song, W.; Zhong, N.; Wu, Y.; Fu, X. Improving Crop Nitrogen Use Efficiency Toward Sustainable Green Revolution. Annu. Rev. Plant Biol. 2022, 73, 523–551. [Google Scholar] [CrossRef]
- Li, S.; Tian, Y.; Wu, K.; Ye, Y.; Yu, J.; Zhang, J.; Liu, Q.; Hu, M.; Li, H.; Tong, Y.; et al. Modulating plant growth-metabolism coordination for sustainable agriculture. Nature 2018, 560, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Wang, W.; Ou, S.; Tang, J.; Li, H.; Che, R.; Zhang, Z.; Chai, X.; Wang, H.; Wang, Y.; et al. Variation in NRT1.1B contributes to nitrate-use divergence between rice subspecies. Nat. Genet. 2015, 47, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, Y.-X.; Zhang, N.; Hu, B.; Jin, T.; Xu, H.; Qin, Y.; Yan, P.; Zhang, X.; Guo, X.; et al. NRT1.1B is associated with root microbiota composition and nitrogen use in field-grown rice. Nat. Biotechnol. 2019, 37, 676–684. [Google Scholar] [CrossRef]
- Tang, W.; Ye, J.; Yao, X.; Zhao, P.; Xuan, W.; Tian, Y.; Zhang, Y.; Xu, S.; An, H.; Chen, G.; et al. Genome-wide associated study identifies NAC42-activated nitrate transporter conferring high nitrogen use efficiency in rice. Nat. Commun. 2019, 10, 5279. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, H.; Jiang, Z.; Wang, W.; Xu, R.; Wang, Q.; Zhang, Z.; Li, A.; Liang, Y.; Ou, S.; et al. Genomic basis of geographical adaptation to soil nitrogen in rice. Nature 2021, 590, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Li, X.; Lu, Z.; Zhang, H.; Ye, X.; Zhou, Y.; Li, J.; Yan, Y.; Pei, H.; Duan, F.; et al. A transcriptional regulator that boosts grain yields and shortens the growth duration of rice. Science 2022, 377, eabi8455. [Google Scholar] [CrossRef] [PubMed]
- Yano, K.; Yamamoto, E.; Aya, K.; Takeuchi, H.; Lo, P.; Hu, L.; Yamasaki, M.; Yoshida, S.; Kitano, H.; Hirano, K.; et al. Genome-wide association study using whole-genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nat. Genet. 2016, 48, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, J.; Yan, Y.; Liu, W.; Zhang, W.; Gao, L.; Tian, Y. Knock-Down of CsNRT2.1, a Cucumber Nitrate Transporter, Reduces Nitrate Uptake, Root length, and Lateral Root Number at Low External Nitrate Concentration. Front. Plant Sci. 2018, 9, 722. [Google Scholar] [CrossRef] [Green Version]
- Balding, D.J. A tutorial on statistical methods for population association studies. Nat. Rev. Genet. 2006, 7, 781–791. [Google Scholar] [CrossRef]
- Gupta, P.K.; Kulwal, P.L.; Jaiswal, V. Association mapping in plants in the post-GWAS genomics era. In Advances in Genetics; Elsevier: Amsterdam, The Netherlands, 2019; Volume 104, pp. 75–154. ISBN 978-0-12-817161-5. [Google Scholar]
- Tibbs Cortes, L.; Zhang, Z.; Yu, J. Status and prospects of genome-wide association studies in plants. Plant Genome 2021, 14, e20077. [Google Scholar] [CrossRef]
- Yasir, M.; Kanwal, H.H.; Hussain, Q.; Riaz, M.W.; Sajjad, M.; Rong, J.; Jiang, Y. Status and prospects of genome-wide association studies in cotton. Front. Plant Sci. 2022, 13, 1019347. [Google Scholar] [CrossRef]
- Xiao, Y.; Liu, H.; Wu, L.; Warburton, M.; Yan, J. Genome-wide Association Studies in Maize: Praise and Stargaze. Mol. Plant 2017, 10, 359–374. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.B. Laboratory Guide for Conducting Soil Tests and Plant Analysis, 1st ed.; CRC Press: Boca Raton, FL, USA, 2001; ISBN 978-0-429-13211-7. [Google Scholar]
- Tiwari, J.K.; Buckseth, T.; Devi, S.; Varshney, S.; Sahu, S.; Patil, V.U.; Zinta, R.; Ali, N.; Moudgil, V.; Singh, R.K.; et al. Physiological and genome-wide RNA-sequencing analyses identify candidate genes in a nitrogen-use efficient potato cv. Kufri Gaurav. Plant Physiol. Biochem. 2020, 154, 171–183. [Google Scholar] [CrossRef]
- Liang, J.; Chen, X.; Guo, P.; Ren, H.; Xie, Z.; Zhang, Z.; Zhen, A. Grafting improves nitrogen-use efficiency by regulating the nitrogen uptake and metabolism under low-nitrate conditions in cucumber. Sci. Hortic. 2021, 289, 110454. [Google Scholar] [CrossRef]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R Package for Multivariate Analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Van der Auwera, G.A.; O’Connor, B.D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra; O’Reilly Media: Newton, MA, USA, 2020; ISBN 978-1-4919-7519-0. [Google Scholar]
- Li, Q.; Li, H.; Huang, W.; Xu, Y.; Zhou, Q.; Wang, S.; Ruan, J.; Huang, S.; Zhang, Z. A chromosome-scale genome assembly of cucumber (Cucumis sativus L.). GigaScience 2019, 8, giz072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, J.; Liu, X.; Shen, D.; Miao, H.; Xie, B.; Li, X.; Zeng, P.; Wang, S.; Shang, Y.; Gu, X.; et al. A genomic variation map provides insights into the genetic basis of cucumber domestication and diversity. Nat Genet 2013, 45, 1510–1515. [Google Scholar] [CrossRef]
- Huang, M.; Liu, X.; Zhou, Y.; Summers, R.M.; Zhang, Z. BLINK: A package for the next level of genome-wide association studies with both individuals and markers in the millions. GigaScience 2019, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhang, Z. GAPIT Version 3: Boosting Power and Accuracy for Genomic Association and Prediction. Genom. Proteom. Bioinform. 2021, 19, 629–640. [Google Scholar] [CrossRef]
- Dong, S.-S.; He, W.-M.; Ji, J.-J.; Zhang, C.; Guo, Y.; Yang, T.-L. LDBlockShow: A fast and convenient tool for visualizing linkage disequilibrium and haplotype blocks based on variant call format files. Brief. Bioinform. 2021, 22, bbaa227. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Bork, P. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Shi, Z.; Qie, Q.; Gao, J.; Wang, X.; Han, Y. CandiHap: A toolkit for haplotype analysis for sequence of samples and fast identification of candidate causal gene(s) in genome-wide association study. bioRxiv 2020. [Google Scholar] [CrossRef]
- Liu, W.; Xie, Y.; Ma, J.; Luo, X.; Nie, P.; Zuo, Z.; Lahrmann, U.; Zhao, Q.; Zheng, Y.; Zhao, Y.; et al. IBS: An illustrator for the presentation and visualization of biological sequences. Bioinformatics 2015, 31, 3359–3361. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Shi, C.; Fu, W.; Gu, X.; Qi, Z.; Xu, W.; Xia, G. Arabidopsis MED18 Interaction With RNA Pol IV and V Subunit NRPD2a in Transcriptional Regulation of Plant Immune Responses. Front. Plant Sci. 2021, 12, 692036. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, M.S.; Shankar, R.; Garg, R.; Jain, M. Bisulphite sequencing reveals dynamic DNA methylation under desiccation and salinity stresses in rice cultivars. Genomics 2020, 112, 3537–3548. [Google Scholar] [CrossRef] [PubMed]
- Van Dooren, T.J.M.; Silveira, A.B.; Gilbault, E.; Jiménez-Gómez, J.M.; Martin, A.; Bach, L.; Tisné, S.; Quadrana, L.; Loudet, O.; Colot, V. Mild drought in the vegetative stage induces phenotypic, gene expression, and DNA methylation plasticity in Arabidopsis but no transgenerational effects. J. Exp. Bot. 2020, 71, 3588–3602. [Google Scholar] [CrossRef]
- Ma, Y.; Min, L.; Wang, M.; Wang, C.; Zhao, Y.; Li, Y.; Fang, Q.; Wu, Y.; Xie, S.; Ding, Y.; et al. Disrupted Genome Methylation in Response to High Temperature Has Distinct Affects on Microspore Abortion and Anther Indehiscence. Plant Cell 2018, 30, 1387–1403. [Google Scholar] [CrossRef] [Green Version]
- Tsay, Y.F.; Schroeder, J.I.; Feldmann, K.A.; Crawford, N.M. The herbicide sensitivity gene CHL1 of Arabidopsis encodes a nitrate-inducible nitrate transporter. Cell 1993, 72, 705–713. [Google Scholar] [CrossRef]
- Naulin, P.A.; Armijo, G.I.; Vega, A.S.; Tamayo, K.P.; Gras, D.E.; de la Cruz, J.; Gutiérrez, R.A. Nitrate Induction of Primary Root Growth Requires Cytokinin Signaling in Arabidopsis thaliana. Plant Cell Physiol. 2020, 61, 342–352. [Google Scholar] [CrossRef]
- Liu, K.-H.; Niu, Y.; Konishi, M.; Wu, Y.; Du, H.; Sun Chung, H.; Li, L.; Boudsocq, M.; McCormack, M.; Maekawa, S.; et al. Discovery of nitrate-CPK-NLP signalling in central nutrient-growth networks. Nature 2017, 545, 311–316. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Mogami, J.; Yamaguchi-Shinozaki, K. ABA-dependent and ABA-independent signaling in response to osmotic stress in plants. Curr. Opin. Plant Biol. 2014, 21, 133–139. [Google Scholar] [CrossRef]
- Belda-Palazón, B.; Adamo, M.; Valerio, C.; Ferreira, L.J.; Confraria, A.; Reis-Barata, D.; Rodrigues, A.; Meyer, C.; Rodriguez, P.L.; Baena-González, E. A dual function of SnRK2 kinases in the regulation of SnRK1 and plant growth. Nat. Plants 2020, 6, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-H.; Wan, S.-Q.; Wang, W.-D.; Chen, J.-F.; Huang, L.-L.; Duan, M.-S.; Yu, Y.-B. Genome-wide identification and characterization of the CsSnRK2 family in Camellia sinensis. Plant Physiol. Biochem. 2018, 132, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wan, S.; Liu, X.; He, J.; Cheng, L.; Duan, M.; Liu, H.; Wang, W.; Yu, Y. Overexpression of CsSnRK2.5 increases tolerance to drought stress in transgenic Arabidopsis. Plant Physiol. Biochem. 2020, 150, 162–170. [Google Scholar] [CrossRef]
- Song, X.; Yu, X.; Hori, C.; Demura, T.; Ohtani, M.; Zhuge, Q. Heterologous Overexpression of Poplar SnRK2 Genes Enhanced Salt Stress Tolerance in Arabidopsis thaliana. Front. Plant Sci. 2016, 7, 612. [Google Scholar] [CrossRef] [Green Version]
- Peng, M.; Bi, Y.-M.; Zhu, T.; Rothstein, S.J. Genome-wide analysis of Arabidopsis responsive transcriptome to nitrogen limitation and its regulation by the ubiquitin ligase gene NLA. Plant Mol. Biol. 2007, 65, 775–797. [Google Scholar] [CrossRef] [PubMed]
- Komarova, N.Y.; Thor, K.; Gubler, A.; Meier, S.; Dietrich, D.; Weichert, A.; Suter Grotemeyer, M.; Tegeder, M.; Rentsch, D. AtPTR1 and AtPTR5 Transport Dipeptides in Planta. Plant Physiol. 2008, 148, 856–869. [Google Scholar] [CrossRef] [PubMed]
- Wen, B.; Xiao, W.; Mu, Q.; Li, D.; Chen, X.; Wu, H.; Li, L.; Peng, F. How does nitrate regulate plant senescence? Plant Physiol. Biochem. 2020, 157, 60–69. [Google Scholar] [CrossRef]
- Luo, L.; Zhang, Y.; Xu, G. How does nitrogen shape plant architecture? J. Exp. Bot. 2020, 71, 4415–4427. [Google Scholar] [CrossRef] [PubMed]
- Mur, L.A.J.; Simpson, C.; Kumari, A.; Gupta, A.K.; Gupta, K.J. Moving nitrogen to the centre of plant defence against pathogens. Ann. Bot. 2017, 119, 703–709. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zou, C.; Gao, X.; Guan, X.; Zhang, Y.; Shi, X.; Chen, X. Nitrate leaching from open-field and greenhouse vegetable systems in China: A meta-analysis. Environ. Sci. Pollut. Res. 2018, 25, 31007–31016. [Google Scholar] [CrossRef]
- Yang, Y.; Gao, S.; Su, Y.; Lin, Z.; Guo, J.; Li, M.; Wang, Z.; Que, Y.; Xu, L. Transcripts and low nitrogen tolerance: Regulatory and metabolic pathways in sugarcane under low nitrogen stress. Environ. Exp. Bot. 2019, 163, 97–111. [Google Scholar] [CrossRef]
- Curci, P.L.; Aiese Cigliano, R.; Zuluaga, D.L.; Janni, M.; Sanseverino, W.; Sonnante, G. Transcriptomic response of durum wheat to nitrogen starvation. Sci. Rep. 2017, 7, 1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandes, N.; Linial, N.; Linial, M. PWAS: Proteome-wide association study-linking genes and phenotypes by functional variation in proteins. Genome Biol. 2020, 21, 173. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Jiang, Y.; Zhong, X.; Cox, N.J.; Liu, C.; Gamazon, E.R. A unified framework for joint-tissue transcriptome-wide association and Mendelian randomization analysis. Nat. Genet. 2020, 52, 1239–1246. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Min | Max | Mean | SD | CV (%) |
|---|---|---|---|---|---|
| DWR | 0.510 | 0.840 | 0.6697 | 0.073 | 10.936 |
| HR | 0.460 | 0.870 | 0.6658 | 0.081 | 12.206 |
| LNC | 0.030 | 0.050 | 0.0347 | 0.004 | 11.297 |
| NAR | 0.090 | 0.260 | 0.1637 | 0.039 | 23.940 |
| NUtER | 0.680 | 1.260 | 0.9655 | 0.136 | 14.111 |
| NUpER | 2.340 | 3.440 | 2.7947 | 0.225 | 8.048 |
| Lead SNP Name | LD Block | Pos | Chr | PVE | Maf | LD Block Range | SNPs Number | Genes Number |
|---|---|---|---|---|---|---|---|---|
| DWR_8554 | 8554 | 3,048,554 | chr3 | 9.84 × 10−6 | 0.31 | 3,046,071–3,048,993 | 27 | 3 |
| HR_1572 | 1572 | 2,541,572 | chr3 | 3.88 × 10−6 | 0.28 | 2,413,014–2,613,011 | 870 | 13 |
| LNC_6476 | 6476 | 9,146,476 | chr3 | 4.34 × 10−6 | 0.26 | 9,132,044–9,168,717 | 123 | 9 |
| LNC_2639 | 2639 | 15,992,639 | chr4 | 7.19 × 10−6 | 0.16 | 15,910,369–16,009,194 | 176 | 6 |
| NAR_1572 | 1572 | 2,541,572 | chr3 | 8.40 × 10−6 | 0.28 | 2,413,014–2,613,011 | 870 | 13 |
| NAR_8554 | 8554 | 3,048,554 | chr3 | 9.06 × 10−6 | 0.31 | 3,046,071–3,048,993 | 27 | 3 |
| NAR_9927 | 9927 | 20,009,927 | chr4 | 3.49 × 10−7 | 0.22 | 19,970,542–20,024,272 | 462 | 5 |
| NUpER_9927 | 9927 | 20,009,927 | chr4 | 1.58 × 10−6 | 0.22 | 19,970,542–20,024,272 | 462 | 5 |
| NUpER_5252 | 5252 | 1,255,252 | chr6 | 8.91 × 10−6 | 0.39 | 1,060,742–1,260,725 | 621 | 25 |
| Gene ID | LD Block | Description | GO Annotation |
|---|---|---|---|
| CsaV3_3G002970 | 1572 | Nitrate transporter | GO:0010167, GO:0015112, GO:0015706, GO:0071705, GO:1901698 |
| CsaV3_3G002990 | 1572 | Adenine phosphoribosyltransferase | GO:0006807, GO:0009308, GO:0034641, GO:0044271, GO:1901564, GO:1901566 |
| CsaV3_3G003050 | 1572 | NAC domain-containing protein | GO:0051171 |
| CsaV3_3G003630 | 8554 | DNA-dependent RNA polymerase catalyzes the transcription of DNA into RNA using the four ribonucleoside triphosphates as substrates | GO:0006807, GO:0034641, GO:1901698, GO:1901699 |
| CsaV3_3G011740 | 6476 | U-box domain-containing protein | GO:0006807, GO:0034641, GO:1901564 |
| CsaV3_3G011750 | 6476 | Prp19/Pso4-like | GO:0006807, GO:0034641, GO:1901564 |
| CsaV3_3G011820 | 6476 | Protein NRT1 PTR FAMILY 5.2-like | GO:0010243, GO:0042886, GO:0042887, GO:0071705, GO:1901698 |
| CsaV3_4G026760 | 2639 | - | GO:0051171, GO:0051173 |
| CsaV3_4G026950 | 2639 | Regulatory-associated protein of TOR (RAPTOR1) | GO:0010243, GO:0051171, GO:0051173, GO:0071417, GO:1901698, GO:1901699 |
| CsaV3_4G030260 | 9927 | Belongs to the protein kinase superfamily (SNRK2.1) | GO:0006807, GO:1901564 |
| CsaV3_6G001670 | 5252 | WUSCHEL-related homeobox (WOX5) | GO:0051171 |
| CsaV3_6G001680 | 5252 | component of the eukaryotic translation initiation factor 3 (eIF-3) complex, which is involved in protein synthesis of a specialized repertoire of mRNAs and, together with other initiation factors, and stimulates binding of mRNA and methionyl-tRNAi to the 40S ribosome. The eIF-3 complex specifically targets and initiates translation of a subset of mRNAs involved in cell proliferation | GO:0006807, GO:0034641, GO:0044271, GO:1901564, GO:1901566 |
| CsaV3_6G001760 | 5252 | Involved in the post-translational conjugation of arginine to the N-terminal aspartate or glutamate of a protein. This arginylation is required for degradation of the protein via the ubiquitin pathway | GO:0006807, GO:1901564, GO:1901565 |
| CsaV3_6G001800 | 5252 | Anthranilate synthase (ASA1) | GO:0006807, GO:0009308, GO:0009309, GO:0034641, GO:0044106, GO:0044271, GO:1901564, GO:1901566 |
| CsaV3_6G001850 | 5252 | NF-X1-type zinc finger protein | GO:0006807, GO:0034641, GO:0044271, GO:0051171, GO:0051172 |
| CsaV3_6G001860 | 5252 | Dual specificity tyrosine-phosphorylation-regulated kinase | GO:0006807, GO:1901564 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, B.; Wei, A.; Tong, X.; Han, Y.; Liu, N.; Chen, Z.; Yang, H.; Wu, H.; Lv, M.; Wang, N.N.; et al. A Genome-Wide Association Study to Identify Novel Candidate Genes Related to Low-Nitrogen Tolerance in Cucumber (Cucumis sativus L.). Genes 2023, 14, 662. https://doi.org/10.3390/genes14030662
Li B, Wei A, Tong X, Han Y, Liu N, Chen Z, Yang H, Wu H, Lv M, Wang NN, et al. A Genome-Wide Association Study to Identify Novel Candidate Genes Related to Low-Nitrogen Tolerance in Cucumber (Cucumis sativus L.). Genes. 2023; 14(3):662. https://doi.org/10.3390/genes14030662
Chicago/Turabian StyleLi, Bowen, Aimin Wei, Xueqiang Tong, Yike Han, Nan Liu, Zhengwu Chen, Hongyu Yang, Huaxiang Wu, Mingjie Lv, Ning Ning Wang, and et al. 2023. "A Genome-Wide Association Study to Identify Novel Candidate Genes Related to Low-Nitrogen Tolerance in Cucumber (Cucumis sativus L.)" Genes 14, no. 3: 662. https://doi.org/10.3390/genes14030662
APA StyleLi, B., Wei, A., Tong, X., Han, Y., Liu, N., Chen, Z., Yang, H., Wu, H., Lv, M., Wang, N. N., & Du, S. (2023). A Genome-Wide Association Study to Identify Novel Candidate Genes Related to Low-Nitrogen Tolerance in Cucumber (Cucumis sativus L.). Genes, 14(3), 662. https://doi.org/10.3390/genes14030662