
Molecular Epidemiology and Diversity of SARS-CoV-2 in Ethiopia, 2020–2022

 , , , , , , , , , , add
Show full author list
, , , , , , , , , , add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Considerations
2.2. Study Design, Period, and Setting
2.3. Quality Assurance
2.4. RNA-Extraction and Next-Generation Sequencing
2.5. Sample Preparation
2.6. Genome Sequencing
2.6.1. Genome Sequencing Using Illumina Sequencing Technologies
Tiling-Based Polymerase Chain Reaction
2.6.2. Genome Sequencing Using Oxford Nanopore Technologies
2.6.3. Metadata Management
2.6.4. Sequence Assembly, Alignment, and Phylogenetic Analysis
2.6.5. Data Analysis and Visualization
3. Results
3.1. Socio-Demographic Characteristics of the Study Participants
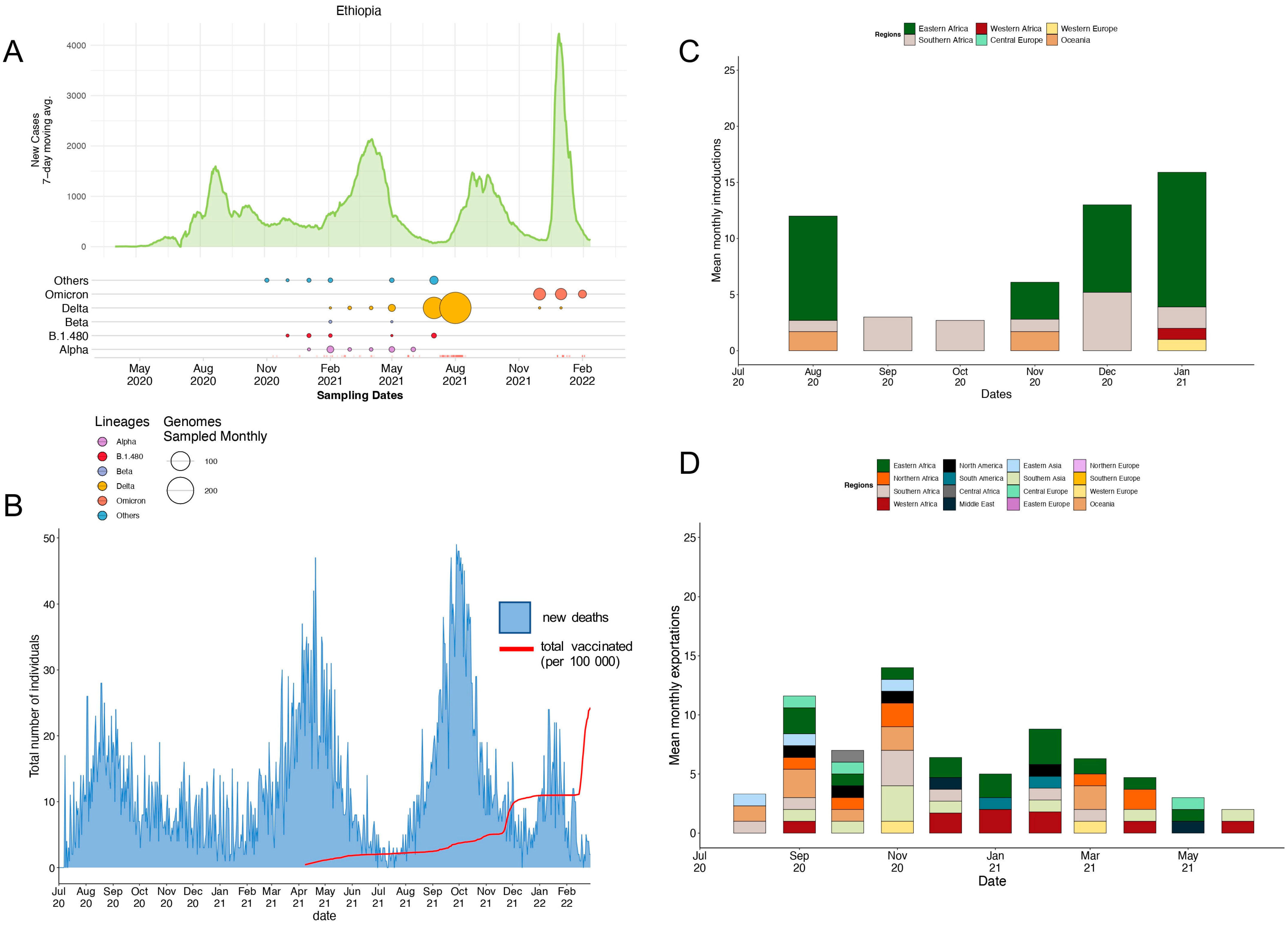
3.2. Local Epidemic Dynamics
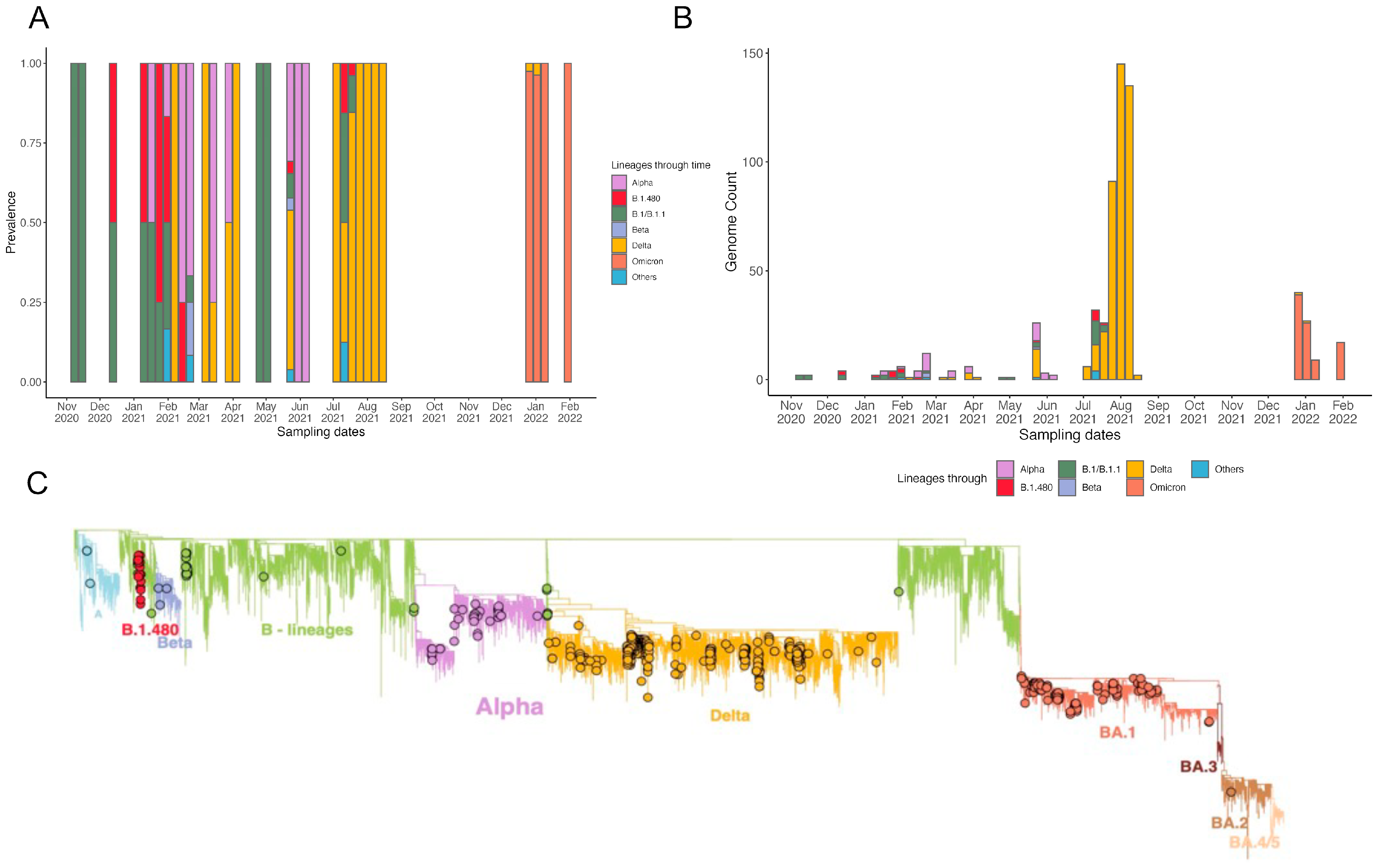
3.3. Phylogenetic Reconstruction and Variant Detection in Ethiopia
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AAPHREML | Addis Ababa Public Health Research and Emergency Management Laboratory |
| AAU | Addis Ababa University |
| ACDC | Africa Center for Disease Prevention and Control |
| CERI | Centre for Epidemic Response and Innovation |
| CI | Confidence interval |
| CT | Cycle threshold |
| GISAID | Global Initiative on Sharing All Influenza Data |
| KRISP | KwaZulu-Natal Research Innovation and Sequencing Platform |
| NAAT | Nucleic acid amplification tests |
| PCR | Polymerase chain reaction |
| RNA | Ribose nucleic acid |
| RT-PCR | Reverse transcriptase polymerase chain reaction |
| SARS-CoV-2 | Severe acute respiratory syndrome novel coronavirus-2 |
| VOCs | Variants of concern |
| VTM | Virus transport medium |
| WHO | World Health Organization |
| SDG | Sustainable development goal |
| COVID-19 | Coronavirus disease 2019 |
References
- University Johns Hopkins. Worldometers COVID-19 Daily Update. 2022. Available online: https://www.worldometers.info/coronavirus/ (accessed on 20 January 2023).
- World Health Organization. World Health Organization Best Practices for the Naming of New Human Infectious Diseases. 2020. Available online: https://www.who.int/publications/i/item/WHO-HSE-FOS-15.1 (accessed on 20 January 2023).
- Bouba, Y.; Tsinda, E.K.; Fonkou, M.D.M.; Mmbando, G.S.; Bragazzi, N.L.; Kong, J.D. The Determinants of the Low COVID-19 Transmission and Mortality Rates in Africa: A Cross-Country Analysis. Front. Public Health 2021, 9, 751197. [Google Scholar] [CrossRef]
- Zerfu, T.A.; Tareke, A.A. What could be the potential reasons for relatively low coronavirus disease 2019 (COVID-19) fatality rates in Africa? The case for Ethiopia. J. Glob. Health 2021, 11, 03057. [Google Scholar] [CrossRef] [PubMed]
- Amadu, I.; Ahinkorah, B.O.; Afitiri, A.-R.; Seidu, A.-A.; Ameyaw, E.K.; Hagan, J.E., Jr.; Duku, E.; Aram, S.A. Assessing sub-regional-specific strengths of healthcare systems associated with COVID-19 prevalence, deaths and recoveries in Africa. PLoS ONE 2021, 16, e0247274. [Google Scholar] [CrossRef]
- Pana, T.A.; Bhattacharya, S.; Gamble, D.T.; Pasdar, Z.; Szlachetka, W.A.; Perdomo-Lampignano, J.A.; Ewers, K.D.; McLernon, D.J.; Myint, P.K. Country-level determinants of the severity of the first global wave of the COVID-19 pandemic: An ecological study. BMJ Open 2021, 11, e042034. [Google Scholar] [CrossRef]
- Adepoju, P. Africa’s struggle with inadequate COVID-19 testing. Lancet Microbe 2020, 1, e12. [Google Scholar] [CrossRef]
- Kobia, F.; Gitaka, J. COVID-19: Are Africa’s diagnostic challenges blunting response effectiveness? AAS Open Res. 2020, 3, 4. [Google Scholar] [CrossRef]
- Wilkinson, E.; Giovanetti, M.; Tegally, H.; San, J.E.; Lessells, R.; Cuadros, D.; Martin, D.P.; Rasmussen, D.A.; Zekri, A.-R.N.; Sangare, A.K.; et al. A year of genomic surveillance reveals how the SARS-CoV-2 pandemic unfolded in Africa. Science 2021, 374, 423–431. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef]
- Tegally, H.; Moir, M.; Everatt, J.; Giovanetti, M.; Scheepers, C.; Wilkinson, E.; Subramoney, K.; Makatini, Z.; Moyo, S.; Amoako, D.G.; et al. Emergence of SARS-CoV-2 Omicron lineages BA.4 and BA.5 in South Africa. Nat. Med. 2022, 28, 1785–1790. [Google Scholar] [CrossRef] [PubMed]
- Bugembe, D.L.; Phan, M.V.T.; Ssewanyana, I.; Semanda, P.; Nansumba, H.; Dhaala, B.; Nabadda, S.; O’Toole, N.; Rambaut, A.; Kaleebu, P.; et al. Emergence and spread of a SARS-CoV-2 lineage A variant (A.23.1) with altered spike protein in Uganda. Nat. Microbiol. 2021, 6, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Olawoye, I.B.; Oluniyi, P.E.; Oguzie, J.U.; Uwanibe, J.N.; Kayode, T.A.; Olumade, T.J.; Ajogbasile, F.V.; Parker, E.; Eromon, P.E.; Abechi, P.; et al. Emergence and spread of two SARS-CoV-2 variants of interest in Nigeria. Nat. Commun. 2023, 14, 811. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, T.; Lutucuta, S.; Nkengasong, J.; Morais, J.; Paixão, J.P.; Neto, Z.; Afonso, P.; Miranda, J.; David, K.; Lessells, R.J.; et al. A novel variant of interest of SARS-CoV-2 with multiple spike mutations detected through travel surveillance in Africa. medRxiv 2021, 3, 21254323. [Google Scholar]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—From vision to reality. Eurosurveillance Bull. Eur. Sur Les Mal. Transm. 2017, 22, 30494. [Google Scholar] [CrossRef] [Green Version]
- Ethiopia: The World Bank. Available online: https://www.worldbank.org/en/country/ethiopia/overview (accessed on 25 January 2023).
- Ebssa, W. The promise of regional projects for Africas landlocked countries: Focusing on Ethiopia. Afr. J. Politi-Sci. Int. Relat. 2015, 9, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Persoz, L.; Hakiza, S.; Biru, L.; Girmatsion, L. Impact of COVID-19 on Food Security in Ethiopia. Epidemiologia 2022, 3, 161–178. [Google Scholar] [CrossRef]
- World Health Organization (WHO Africa). First Case of COVID-19 Confirmed in Ethiopia. Available online: https://www.afro.who.int/news/first-case-covid-19-confirmed-ethiopia#:~:text=March%2013%2C%202020%2C%20The%20Federal,in%20China%20in%20December%202019 (accessed on 10 December 2022).
- Lanyero, B.; Edea, Z.A.; Musa, E.O.; Watare, S.H.; Mandalia, M.L.; Livinus, M.C.; Ebrahim, F.K.; Girmay, A.; Bategereza, A.K.; Abayneh, A.; et al. Readiness and early response to COVID-19: Achievements, challenges and lessons learnt in Ethiopia. BMJ Glob. Health 2021, 6, e005581. [Google Scholar] [CrossRef]
- Ethiopian Public Health Institute, EPHI. COVID-19 Pandemic Preparedness and Response in Ethiopia Weekly Bulletin November 2021. Updated 2022. Available online: https://ephi.gov.et/download/pheoc/ (accessed on 20 December 2022).
- Amhare, A.F.; Tao, Y.; Li, R.; Zhang, L. Early and Subsequent Epidemic Characteristics of COVID-19 and Their Impact on the Epidemic Size in Ethiopia. Front. Public Health 2022, 10, 834592. [Google Scholar] [CrossRef]
- Abdella, S.; Riou, S.; Tessema, M.; Assefa, A.; Seifu, A.; Blachman, A.; Abera, A.; Moreno, N.; Irarrazaval, F.; Tollera, G.; et al. Prevalence of SARS-CoV-2 in urban and rural Ethiopia: Randomized household serosurveys reveal level of spread during the first wave of the pandemic. Eclinicalmedicine 2021, 35, 100880. [Google Scholar] [CrossRef]
- Sisay, A.; Abera, A.; Dufera, B.; Endrias, T.; Tasew, G.; Tesfaye, A.; Hartnack, S.; Beyene, D.; Desta, A.F. Diagnostic accuracy of three commercially available one step RT-PCR assays for the detection of SARS-CoV-2 in resource limited settings. PLoS ONE 2022, 17, e0262178. [Google Scholar] [CrossRef] [PubMed]
- Tshiabuila, D.; Giandhari, J.; Pillay, S.; Ramphal, U.; Ramphal, Y.; Maharaj, A.; Anyaneji, U.J.; Naidoo, Y.; Tegally, H.; San, E.J.; et al. Comparison of SARS-CoV-2 sequencing using the ONT GridION and the Illumina MiSeq. BMC Genom. 2022, 23, 319. [Google Scholar] [CrossRef] [PubMed]
- Pembaur, A.; Sallard, E.; Weil, P.P.; Ortelt, J.; Ahmad-Nejad, P.; Postberg, J. Simplified Point-of-Care Full SARS-CoV-2 Genome Sequencing Using Nanopore Technology. Microorganisms 2021, 9, 2598. [Google Scholar] [CrossRef]
- Vilsker, M.; Moosa, Y.; Nooij, S.; Fonseca, V.; Ghysens, Y.; Dumon, K.; Pauwels, R.; Alcantara, L.C.; Vanden Eynden, E.; Vandamme, A.-M.; et al. Genome Detective: An automated system for virus identification from high-throughput sequencing data. Bioinformatics 2019, 35, 871–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleemput, S.; Dumon, W.; Fonseca, V.; Abdool Karim, W.; Giovanetti, M.; Alcantara, L.C.; Deforche, K.; De Oliveira, T. Genome Detective Coronavirus Typing Tool for rapid identification and characterization of novel coronavirus genomes. Bioinformatics 2020, 36, 3552–3555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nextclade. Next Clade. 2021. Available online: https://clades.nextstrain.org/ (accessed on 15 December 2022).
- Aksamentov, I.; Neher, R. Nextalign: Viral Genome Reference Alignment. Available online: https://github.com/neherlab/nextalign (accessed on 16 January 2023).
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Gatto, L.; Catanzaro, D.; Milinkovitch, M. Assessing the Applicability of the GTR Nucleotide Substitution Model through Simulations. Evol. Bioinform. 2007, 2, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y. GGTREE: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Tegally, H.; San, J.E.; Cotten, M.; Moir, M.; Tegomoh, B.; Mboowa, G.; Martin, D.P.; Baxter, C.; Lambisia, A.W.; Diallo, A.; et al. The evolving SARS-CoV-2 epidemic in Africa: Insights from rapidly expanding genomic surveillance. Science 2022, 378, eabq5358. [Google Scholar] [CrossRef]
- Ellingjord-Dale, M.; Kalleberg, K.T.; Istre, M.S.; Nygaard, A.B.; Brunvoll, S.H.; Eggesbø, L.M.; Dahl, J.A.; Kjetland, E.F.; Ursin, G.; Søraas, A. The use of public transport and contraction of SARS-CoV-2 in a large prospective cohort in Norway. BMC Infect. Dis. 2022, 22, 252. [Google Scholar] [CrossRef]
- Mekuriaw, W.; Kinde, S.; Kindu, B.; Mulualem, Y.; Hailu, G.; Gebresilassie, A.; Sisay, C.; Bekele, F.; Amare, H.; Wossen, M.; et al. Epidemiological, Entomological, and Climatological Investigation of the 2019 Dengue Fever Outbreak in Gewane District, Afar Region, North-East Ethiopia. Insects 2022, 13, 1066. [Google Scholar] [CrossRef] [PubMed]
- Tsegaye, M.M.; Beyene, B.; Ayele, W.; Abebe, A.; Tareke, I.; Sall, A.; Yactayo, S.; Shibeshi, M.E.; Staples, E.; Belay, D.; et al. Sero-prevalence of yellow fever and related Flavi viruses in Ethiopia: A public health perspective. BMC Public Health 2018, 18, 1011. [Google Scholar] [CrossRef] [Green Version]
- Tessema, G.A.; Kinfu, Y.; Dachew, B.A.; Tesema, A.G.; Assefa, Y.; Alene, K.A.; Aregay, A.F.; Ayalew, M.B.; Bezabhe, W.M.; Bali, A.G.; et al. The COVID-19 pandemic and healthcare systems in Africa: A scoping review of preparedness, impact and response. BMJ Glob. Health 2021, 6, e007179. [Google Scholar] [CrossRef] [PubMed]
- Abagero, A.; Ragazzoni, L.; Hubloue, I.; Barone-Adesi, F.; Lamine, H.; Addissie, A.; Della Corte, F.; Valente, M. A Review of COVID-19 Response Challenges in Ethiopia. Int. J. Environ. Res. Public Health 2022, 19, 11070. [Google Scholar] [CrossRef]
- Nigussie, H. The Coronavirus Intervention in Ethiopia and the Challenges for Implementation. Front. Commun. 2021, 6, 562512. [Google Scholar] [CrossRef]
- Ngeh, S.; Vogt, F.; Sikazwe, C.T.; Levy, A.; Pingault, N.M.; Smith, D.W.; Effler, P.V. Travel-associated SARS-CoV-2 transmission documented with whole genome sequencing following a long-haul international flight. J. Travel Med. 2022, 29, taac057. [Google Scholar] [CrossRef]
- Gelanew, T.; Seyoum, B.; Mulu, A.; Mihret, A.; Abebe, M.; Wassie, L.; Gelaw, B.; Sorsa, A.; Merid, Y.; Muchie, Y.; et al. High seroprevalence of anti-SARS-CoV-2 antibodies among Ethiopian healthcare workers. BMC Infect. Dis. 2022, 22, 261. [Google Scholar] [CrossRef]
- Gudina, E.K.; Ali, S.; Girma, E.; Gize, A.; Tegene, B.; Hundie, G.B.; Sime, W.T.; Ambachew, R.; Gebreyohanns, A.; Bekele, M.; et al. Seroepidemiology and model-based prediction of SARS-CoV-2 in Ethiopia: Longitudinal cohort study among front-line hospital workers and communities. Lancet Glob. Health 2021, 9, e1517–e1527. [Google Scholar] [CrossRef]
- COVID-19 Vaccine Tracker. Available online: https://www.reuters.com/graphics/world-coronavirus-tracker-and-maps/vaccination-rollout-and-access/ (accessed on 15 December 2022).
- Wilkinson, S.A.J.; Richter, A.; Casey, A.; Osman, H.; Mirza, J.D.; Stockton, J.; Quick, J.; Ratcliffe, L.; Sparks, N.; Loman, N.J.; et al. Recurrent SARS-CoV-2 mutations in immunodeficient patients. Virus Evol. 2022, 8, veac050. [Google Scholar] [CrossRef]
- Weigang, S.; Fuchs, J.; Zimmer, G.; Schnepf, D.; Kern, L.; Beer, J.; Luxenburger, H.; Ankerhold, J.; Falcone, V.; Kemming, J.; et al. Within-host evolution of SARS-CoV-2 in an immunosuppressed COVID-19 patient as a source of immune escape variants. Nat. Commun. 2021, 12, 6405. [Google Scholar] [CrossRef] [PubMed]
- Moghadas, S.M.; Vilches, T.N.; Zhang, K.; Wells, C.R.; Shoukat, A.; Singer, B.H.; Meyers, L.A.; Neuzil, K.M.; Langley, J.M.; Fitzpatrick, M.C.; et al. The Impact of Vaccination on Coronavirus Disease 2019 (COVID-19) Outbreaks in the United States. Clin. Infect. Dis. 2021, 73, 2257–2264. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Consortium, C.-G.U.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Liu, Y.; Rocklöv, J. The reproductive number of the Delta variant of SARS-CoV-2 is far higher compared to the ancestral SARS-CoV-2 virus. J. Travel Med. 2021, 28, taab124. [Google Scholar] [CrossRef]
- Thakur, S.; Sasi, S.; Pillai, S.G.; Nag, A.; Shukla, D.; Singhal, R.; Phalke, S.; Velu, G.S.K. SARS-CoV-2 Mutations and Their Impact on Diagnostics, Therapeutics and Vaccines. Front. Med. 2022, 9, 815389. [Google Scholar] [CrossRef]
- Dhama, K.; Nainu, F.; Frediansyah, A.; Yatoo, M.I.; Mohapatra, R.K.; Chakraborty, S.; Zhou, H.; Islam, M.R.; Mamada, S.S.; Harapan, H.; et al. Global emerging Omicron variant of SARS-CoV-2: Impacts, challenges and strategies. J. Infect. Public Health 2023, 16, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhi, H.; Teng, Y. The outbreak of SARS-CoV-2 Omicron lineages, immune escape, and vaccine effectivity. J. Med. Virol. 2022, 95, e28138. [Google Scholar] [CrossRef] [PubMed]
- Corriero, A.; Ribezzi, M.; Mele, F.; Angrisani, C.; Romaniello, F.; Daleno, A.; Loconsole, D.; Centrone, F.; Chironna, M.; Brienza, N. COVID-19 Variants in Critically Ill Patients: A Comparison of the Delta and Omicron Variant Profiles. Infect. Dis. Rep. 2022, 14, 492–500. [Google Scholar] [CrossRef]
- Dubey, A.; Choudhary, S.; Kumar, P.; Tomar, S. Emerging SARS-CoV-2 Variants: Genetic Variability and Clinical Implications. Curr. Microbiol. 2022, 79, 20. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Variable | Class | Frequency (%) | Variant (VOC) | |||
|---|---|---|---|---|---|---|
| Alpha | Beta | Delta | Omicron | |||
| Sex | Female | 181 (51.3) | 10 | 2 | 99 | 57 |
| Male | 172 (48.7) | 7 | 1 | 102 | 46 | |
| Age range (years) | 1–20 | 40 (11.3) | 7 | 1 | 19 | 8 |
| 21–30 | 121 (34.3) | 1 | 1 | 80 | 25 | |
| 31–40 | 84 (23.8) | 5 | 1 | 43 | 30 | |
| 41–50 | 41 (11.6) | 2 | 0 | 25 | 12 | |
| 51–60 | 21 (10.2) | 2 | 0 | 18 | 15 | |
| >60 | 31 (8.8) | 0 | 0 | 16 | 13 | |
| Reason for testing (during sampling) | Suspect | 107 (30.3) | 6 | 1 | 133 | 2 |
| Contacts of confirmed cases | 79 (22.4) | 8 | 2 | 45 | 1 | |
| Community Surveillance | 167 (47.3) | 3 | 0 | 23 | 100 | |
| Clinical status of the clients while sampling | Asymptomatic * | 53 (15) | 3 | 0 | 37 | 5 |
| Mild | 273 (77.3) | 14 | 3 | 142 | 94 | |
| Severe | 27 (7.7) | 0 | 0 | 22 | 4 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sisay, A.; Tshiabuila, D.; van Wyk, S.; Tesfaye, A.; Mboowa, G.; Oyola, S.O.; Tesema, S.K.; Baxter, C.; Martin, D.; Lessells, R.; et al. Molecular Epidemiology and Diversity of SARS-CoV-2 in Ethiopia, 2020–2022. Genes 2023, 14, 705. https://doi.org/10.3390/genes14030705
Sisay A, Tshiabuila D, van Wyk S, Tesfaye A, Mboowa G, Oyola SO, Tesema SK, Baxter C, Martin D, Lessells R, et al. Molecular Epidemiology and Diversity of SARS-CoV-2 in Ethiopia, 2020–2022. Genes. 2023; 14(3):705. https://doi.org/10.3390/genes14030705
Chicago/Turabian StyleSisay, Abay, Derek Tshiabuila, Stephanie van Wyk, Abraham Tesfaye, Gerald Mboowa, Samuel O. Oyola, Sofonias Kifle Tesema, Cheryl Baxter, Darren Martin, Richard Lessells, and et al. 2023. "Molecular Epidemiology and Diversity of SARS-CoV-2 in Ethiopia, 2020–2022" Genes 14, no. 3: 705. https://doi.org/10.3390/genes14030705
APA StyleSisay, A., Tshiabuila, D., van Wyk, S., Tesfaye, A., Mboowa, G., Oyola, S. O., Tesema, S. K., Baxter, C., Martin, D., Lessells, R., Tegally, H., Moir, M., Giandhari, J., Pillay, S., Singh, L., Ramphal, Y., Maharaj, A., Pillay, Y., Maharaj, A., ... San, J. E. (2023). Molecular Epidemiology and Diversity of SARS-CoV-2 in Ethiopia, 2020–2022. Genes, 14(3), 705. https://doi.org/10.3390/genes14030705