
Role of Microbiota-Modified Bile Acids in the Regulation of Intracellular Organelles and Neurodegenerative Diseases


{kind=link}
{kind=link}
Abstract
1. Introduction
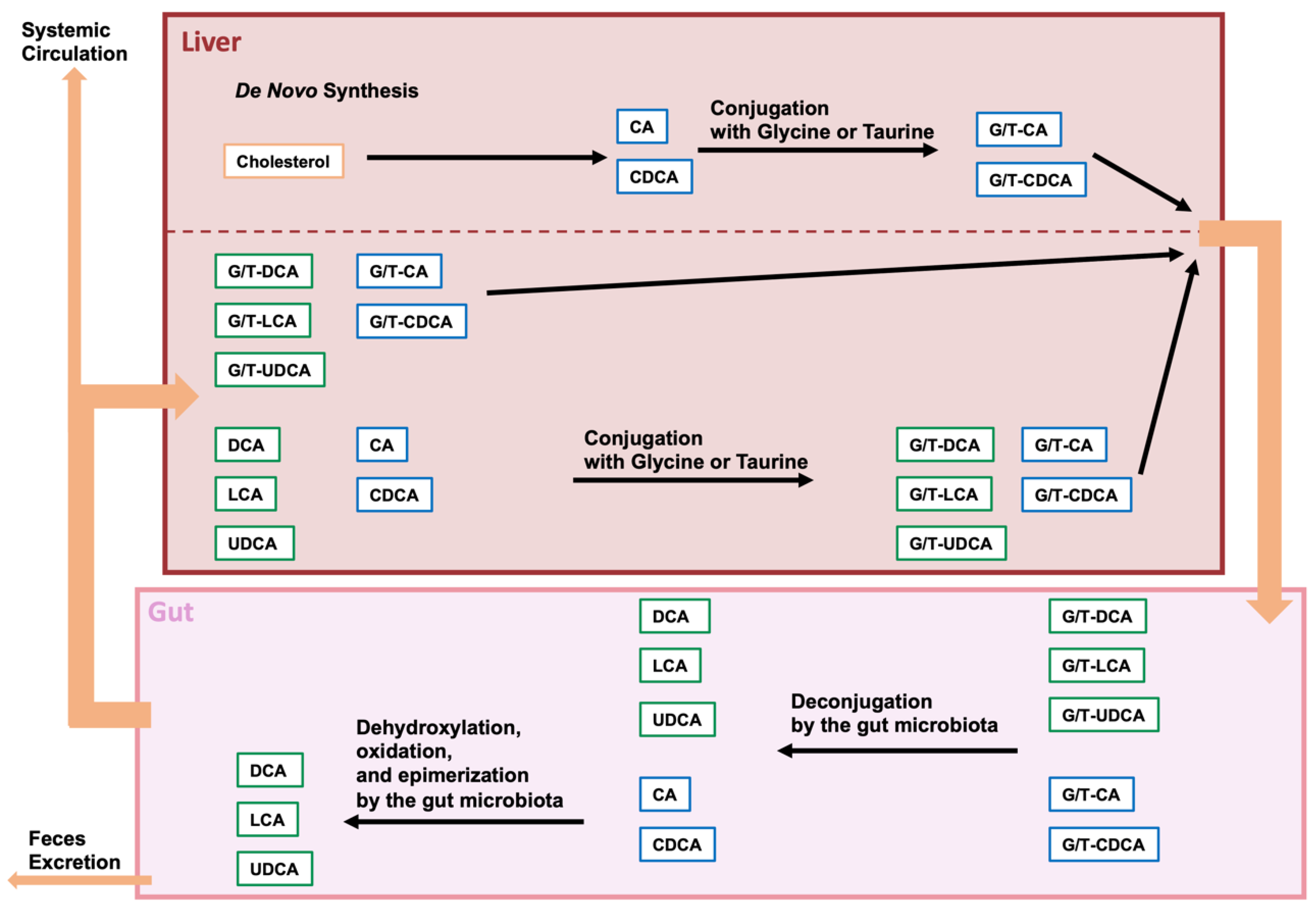
2. BAs and Gut Microbiota
2.1. Bile Acid Modification by the Gut Microbiota
2.2. Analysis of the Composition of the Gut Microbiota and Bacterial Species with Enzymes That Modify BAs
3. BAs as Regulatory Modulators
3.1. BAs as Regulatory Molecules of Nuclear and Cell-Surface Receptors
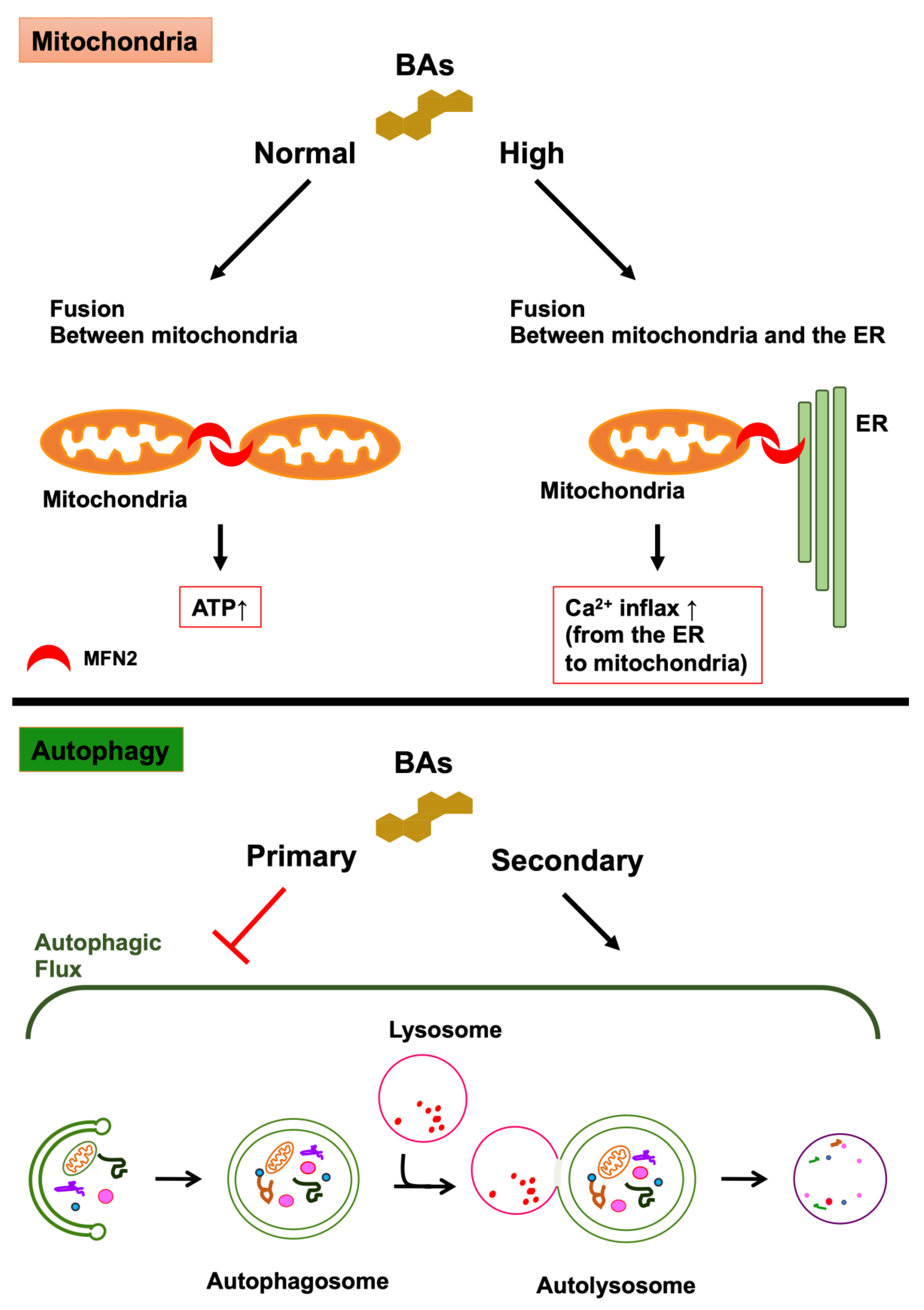
3.2. BAs as Regulatory Molecules of Intracellular Organelle
4. Microbiota-Modified BAs in Neurodegenerative Diseases
4.1. Alzheimer’s Disease
4.2. Parkinson’s Disease
4.3. Huntington’s Disease
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Di Ciaula, A.; Garruti, G.; Baccetto, R.L.; Molina-Molina, E.; Bonfrate, L.; Wang, D.Q.-H.; Portincasa, P. Bile Acid Physiology. Ann. Hepatol. 2017, 16, S4–S14. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chiang, J.Y.L. Bile Acid Signaling in Metabolic Disease and Drug Therapy. Pharmacol. Rev. 2014, 66, 948–983. [Google Scholar] [CrossRef]
- Vallim, T.Q.D.A.; Tarling, E.J.; Edwards, P.A. Pleiotropic Roles of Bile Acids in Metabolism. Cell Metab. 2013, 17, 657–669. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Larabi, A.B.; Masson, H.L.P.; Bäumler, A.J. Bile acids as modulators of gut microbiota composition and function. Gut Microbes 2023, 15, 2172671. [Google Scholar] [CrossRef]
- Perino, A.; Demagny, H.; Velazquez-Villegas, L.A.; Schoonjans, K. Molecular physiology of bile acid signaling in health, disease, and aging. Physiol. Rev. 2021, 101, 683–731. [Google Scholar] [CrossRef]
- Kiriyama, Y.; Nochi, H. Physiological Role of Bile Acids Modified by the Gut Microbiome. Microorganisms 2021, 10, 68. [Google Scholar] [CrossRef]
- Grant, S.; DeMorrow, S. Bile Acid Signaling in Neurodegenerative and Neurological Disorders. Int. J. Mol. Sci. 2020, 21, 5982. [Google Scholar] [CrossRef]
- McMillin, M.; DeMorrow, S. Effects of bile acids on neurological function and disease. FASEB J. 2016, 30, 3658–3668. [Google Scholar] [CrossRef]
- Che, Y.; Xu, W.; Ding, C.; He, T.; Xu, X.; Shuai, Y.; Huang, H.; Wu, J.; Wang, Y.; Wang, C.; et al. Bile acids target mitofusin 2 to differentially regulate innate immunity in physiological versus cholestatic conditions. Cell Rep. 2023, 42, 112011. [Google Scholar] [CrossRef]
- Rolo, A.P.; Palmeira, C.M.; Holy, J.M.; Wallace, K.B. Role of Mitochondrial Dysfunction in Combined Bile Acid-Induced Cytotoxicity: The Switch Between Apoptosis and Necrosis. Toxicol. Sci. 2004, 79, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, I.; Gordino, G.; Moreira, S.; Nunes, M.J.; Azevedo, C.; Gama, M.J.; Rodrigues, E.; Rodrigues, C.M.P.; Castro-Caldas, M. Tauroursodeoxycholic Acid Protects Against Mitochondrial Dysfunction and Cell Death via Mitophagy in Human Neuroblastoma Cells. Mol. Neurobiol. 2016, 54, 6107–6119. [Google Scholar] [CrossRef] [PubMed]
- Manley, S.; Ni, H.-M.; Kong, B.; Apte, U.; Guo, G.; Ding, W.-X. Suppression of Autophagic Flux by Bile Acids in Hepatocytes. Toxicol. Sci. 2013, 137, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Roesly, H.B.; Khan, M.R.; Chen, H.D.R.; Hill, K.A.; Narendran, N.; Watts, G.S.; Chen, X.; Dvorak, K. The decreased expression of Beclin-1 correlates with progression to esophageal adenocarcinoma: The role of deoxycholic acid. Am. J. Physiol. Liver Physiol. 2012, 302, G864–G872. [Google Scholar] [CrossRef] [PubMed]
- Panzitt, K.; Jungwirth, E.; Krones, E.; Lee, J.M.; Pollheimer, M.; Thallinger, G.G.; Kolb-Lenz, D.; Xiao, R.; Thorell, A.; Trauner, M.; et al. FXR-dependent Rubicon induction impairs autophagy in models of human cholestasis. J. Hepatol. 2020, 72, 1122–1131. [Google Scholar] [CrossRef]
- Kiriyama, Y.; Nochi, H. The Biosynthesis, Signaling, and Neurological Functions of Bile Acids. Biomolecules 2019, 9, 232. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.W. Fifty years of advances in bile acid synthesis and metabolism. J. Lipid Res. 2009, 50, S120–S125. [Google Scholar] [CrossRef] [PubMed]
- Katafuchi, T.; Makishima, M. Molecular Basis of Bile Acid-FXR-FGF15/19 Signaling Axis. Int. J. Mol. Sci. 2022, 23, 6046. [Google Scholar] [CrossRef]
- Pandak, W.M.; Kakiyama, G. The acidic pathway of bile acid synthesis: Not just an alternative pathway. Liver Res. 2019, 3, 88–98. [Google Scholar] [CrossRef]
- Styles, N.A.; Shonsey, E.M.; Falany, J.L.; Guidry, A.L.; Barnes, S.; Falany, C.N. Carboxy-terminal mutations of bile acid CoA:N-acyltransferase alter activity and substrate specificity. J. Lipid Res. 2016, 57, 1133–1143. [Google Scholar] [CrossRef]
- Jones, B.V.; Begley, M.; Hill, C.; Gahan, C.G.M.; Marchesi, J.R. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc. Natl. Acad. Sci. USA 2008, 105, 13580–13585. [Google Scholar] [CrossRef]
- O’Flaherty, S.; Crawley, A.B.; Theriot, C.M.; Barrangou, R. The Lactobacillus Bile Salt Hydrolase Repertoire Reveals Niche-Specific Adaptation. Msphere 2018, 3, e00140-18. [Google Scholar] [CrossRef] [PubMed]
- Clarke, G.; Sandhu, K.V.; Griffin, B.T.; Dinan, T.G.; Cryan, J.F.; Hyland, N.P. Gut Reactions: Breaking Down Xenobiotic–Microbiome Interactions. Pharmacol. Rev. 2019, 71, 198–224. [Google Scholar] [CrossRef]
- Rivière, A.; Selak, M.; Lantin, D.; Leroy, F.; De Vuyst, L. Bifidobacteria and Butyrate-Producing Colon Bacteria: Importance and Strategies for Their Stimulation in the Human Gut. Front. Microbiol. 2016, 7, 979. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Okpara, E.S.; Hu, W.; Yan, C.; Wang, Y.; Liang, Q.; Chiang, J.Y.L.; Han, S. Interactive Relationships between Intestinal Flora and Bile Acids. Int. J. Mol. Sci. 2022, 23, 8343. [Google Scholar] [CrossRef]
- Begley, M.; Hill, C.; Gahan, C.G.M. Bile Salt Hydrolase Activity in Probiotics. Appl. Environ. Microbiol. 2006, 72, 1729–1738. [Google Scholar] [CrossRef] [PubMed]
- Doden, H.; Ridlon, J. Microbial Hydroxysteroid Dehydrogenases: From Alpha to Omega. Microorganisms 2021, 9, 469. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Pérez, O.; Cruz-Ramón, V.; Chinchilla-López, P.; Méndez-Sánchez, N. The Role of the Gut Microbiota in Bile Acid Metabolism. Ann. Hepatol. 2017, 16, s15–s20. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, S.; Wang, M.; Hu, H.; Yin, J.; Liu, C.; Huang, Y. Gut Microbiota Dysbiosis Associated with Bile Acid Metabolism in Neonatal Cholestasis Disease. Sci. Rep. 2020, 10, 7686. [Google Scholar] [CrossRef]
- Majait, S.; Nieuwdorp, M.; Kemper, M.; Soeters, M. The Black Box Orchestra of Gut Bacteria and Bile Acids: Who Is the Conductor? Int. J. Mol. Sci. 2023, 24, 1816. [Google Scholar] [CrossRef]
- Song, Z.; Cai, Y.; Lao, X.; Wang, X.; Lin, X.; Cui, Y.; Kalavagunta, P.K.; Liao, J.; Jin, L.; Shang, J.; et al. Taxonomic profiling and populational patterns of bacterial bile salt hydrolase (BSH) genes based on worldwide human gut microbiome. Microbiome 2019, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Wise, J.L.; Cummings, B.P. The 7-α-dehydroxylation pathway: An integral component of gut bacterial bile acid metabolism and potential therapeutic target. Front. Microbiol. 2023, 13, 1093420. [Google Scholar] [CrossRef] [PubMed]
- Guzior, D.V.; Quinn, R.A. Review: Microbial transformations of human bile acids. Microbiome 2021, 9, 140. [Google Scholar] [CrossRef]
- Spichak, S.; Bastiaanssen, T.F.; Berding, K.; Vlckova, K.; Clarke, G.; Dinan, T.G.; Cryan, J.F. Mining microbes for mental health: Determining the role of microbial metabolic pathways in human brain health and disease. Neurosci. Biobehav. Rev. 2021, 125, 698–761. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, L.; Cantoni, C.; Rotondo, E.; Galimberti, D. The Gut Microbiome–Brain Crosstalk in Neurodegenerative Diseases. Biomedicines 2022, 10, 1486. [Google Scholar] [CrossRef]
- Jansson, J.K.; Baker, E.S. A multi-omic future for microbiome studies. Nat. Microbiol. 2016, 1, 16049. [Google Scholar] [CrossRef] [PubMed]
- Jovel, J.; Patterson, J.; Wang, W.; Hotte, N.; O’Keefe, S.; Mitchel, T.; Perry, T.; Kao, D.; Mason, A.L.; Madsen, K.L.; et al. Characterization of the Gut Microbiome Using 16S or Shotgun Metagenomics. Front. Microbiol. 2016, 7, 459. [Google Scholar] [CrossRef]
- Laudadio, I.; Fulci, V.; Palone, F.; Stronati, L.; Cucchiara, S.; Carissimi, C. Quantitative Assessment of Shotgun Metagenomics and 16S rDNA Amplicon Sequencing in the Study of Human Gut Microbiome. OMICS J. Integr. Biol. 2018, 22, 248–254. [Google Scholar] [CrossRef]
- D’Argenio, V. Human Microbiome Acquisition and Bioinformatic Challenges in Metagenomic Studies. Int. J. Mol. Sci. 2018, 19, 383. [Google Scholar] [CrossRef] [PubMed]
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 13537. [Google Scholar] [CrossRef]
- Liu, P.; Wu, L.; Peng, G.; Han, Y.; Tang, R.; Ge, J.; Zhang, L.; Jia, L.; Yue, S.; Zhou, K.; et al. Altered microbiomes distinguish Alzheimer’s disease from amnestic mild cognitive impairment and health in a Chinese cohort. Brain Behav. Immun. 2019, 80, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Haran, J.P.; Bhattarai, S.K.; Foley, S.E.; Dutta, P.; Ward, D.V.; Bucci, V.; McCormick, B.A. Alzheimer’s Disease Microbiome Is Associated with Dysregulation of the Anti-Inflammatory P-Glycoprotein Pathway. Mbio 2019, 10, e00632-19. [Google Scholar] [CrossRef] [PubMed]
- Bedarf, J.R.; Hildebrand, F.; Coelho, L.P.; Sunagawa, S.; Bahram, M.; Goeser, F.; Bork, P.; Wüllner, U. Functional implications of microbial and viral gut metagenome changes in early stage L-DOPA-naïve Parkinson’s disease patients. Genome Med. 2017, 9, 39. [Google Scholar] [CrossRef]
- Li, C.; Cui, L.; Yang, Y.; Miao, J.; Zhao, X.; Zhang, J.; Cui, G.; Zhang, Y. Gut Microbiota Differs Between Parkinson’s Disease Patients and Healthy Controls in Northeast China. Front. Mol. Neurosci. 2019, 12, 171. [Google Scholar] [CrossRef]
- Barichella, M.; Severgnini, M.; Cilia, R.; Cassani, E.; Bolliri, C.; Caronni, S.; Ferri, V.; Cancello, R.; Ceccarani, C.; Faierman, S.; et al. Unraveling gut microbiota in Parkinson’s disease and atypical parkinsonism. Mov. Disord. 2018, 34, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Du, G.; Dong, W.; Yang, Q.; Yu, X.; Ma, J.; Gu, W.; Huang, Y. Altered Gut Microbiota Related to Inflammatory Responses in Patients With Huntington’s Disease. Front. Immunol. 2021, 11, 603594. [Google Scholar] [CrossRef] [PubMed]
- Wasser, C.I.; Mercieca, E.-C.; Kong, G.; Hannan, A.J.; McKeown, S.J.; Glikmann-Johnston, Y.; Stout, J.C. Gut dysbiosis in Huntington’s disease: Associations among gut microbiota, cognitive performance and clinical outcomes. Brain Commun. 2020, 2, 110. [Google Scholar] [CrossRef] [PubMed]
- Wallen, Z.D.; Demirkan, A.; Twa, G.; Cohen, G.; Dean, M.N.; Standaert, D.G.; Sampson, T.R.; Payami, H. Metagenomics of Parkinson’s disease implicates the gut microbiome in multiple disease mechanisms. Nat. Commun. 2022, 13, 6958. [Google Scholar] [CrossRef]
- Zhou, M.; Wang, D.; Li, X.; Cao, Y.; Yi, C.; Ocansey, D.K.W.; Zhou, Y.; Mao, F. Farnesoid-X receptor as a therapeutic target for inflammatory bowel disease and colorectal cancer. Front. Pharmacol. 2022, 13, 1016836. [Google Scholar] [CrossRef]
- Biagioli, M.; Fiorucci, S. Bile acid activated receptors: Integrating immune and metabolic regulation in non-alcoholic fatty liver disease. Liver Res. 2021, 5, 119–141. [Google Scholar] [CrossRef]
- Fiorucci, S.; Distrutti, E. Bile Acid-Activated Receptors, Intestinal Microbiota, and the Treatment of Metabolic Disorders. Trends Mol. Med. 2015, 21, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Portincasa, P.; Di Ciaula, A.; Garruti, G.; Vacca, M.; De Angelis, M.; Wang, D.Q.-H. Bile Acids and GPBAR-1: Dynamic Interaction Involving Genes, Environment and Gut Microbiome. Nutrients 2020, 12, 3709. [Google Scholar] [CrossRef]
- Duboc, H.; Taché, Y.; Hofmann, A.F. The bile acid TGR5 membrane receptor: From basic research to clinical application. Dig. Liver Dis. 2014, 46, 302–312. [Google Scholar] [CrossRef]
- Huang, Y.; Thathiah, A. Regulation of neuronal communication by G protein-coupled receptors. FEBS Lett. 2015, 589, 1607–1619. [Google Scholar] [CrossRef]
- Tayebati, S.K. Phospholipid and Lipid Derivatives as Potential Neuroprotective Compounds. Molecules 2018, 23, 2257. [Google Scholar] [CrossRef] [PubMed]
- Healy, L.; Antel, J. Sphingosine-1-Phosphate Receptors in the Central Nervous and Immune Systems. Curr. Drug Targets 2016, 17, 1841–1850. [Google Scholar] [CrossRef]
- Maria, A.A.; Andréas, M.-R.O.; Haider, N.B. Role of Nuclear Receptors in Central Nervous System Development and Associated Diseases. J. Exp. Neurosci. 2015, 9 (Suppl. S2), 93–121. [Google Scholar] [CrossRef]
- Frye, C.A.; Paris, J.J.; A Walf, A.; Rusconi, J.C. Effects and Mechanisms of 3α,5α,-THP on Emotion, Motivation, and Reward Functions Involving Pregnane Xenobiotic Receptor. Front. Neurosci. 2012, 5, 136. [Google Scholar] [CrossRef]
- Thibaut, M.M.; Bindels, L.B. Crosstalk between bile acid-activated receptors and microbiome in entero-hepatic inflammation. Trends Mol. Med. 2022, 28, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Wang, S.; Wang, P.; Tang, C.; Wang, Z.; Chen, L.; Luo, G.; Chen, H.; Liu, Y.; Feng, B.; et al. Bile acids and their receptors in regulation of gut health and diseases. Prog. Lipid Res. 2023, 89, 101210. [Google Scholar] [CrossRef] [PubMed]
- Kiriyama, Y.; Nochi, H. The Function of Autophagy in Neurodegenerative Diseases. Int. J. Mol. Sci. 2015, 16, 26797–26812. [Google Scholar] [CrossRef] [PubMed]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, E.; Musich, P.R.; Lin, F. Mitochondrial dysfunction in neurodegenerative diseases and the potential countermeasure. CNS Neurosci. Ther. 2019, 25, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu-Norberg, H.; Ouchida, A.T.; Norberg, E. The role of mitochondria in metabolism and cell death. Biochem. Biophys. Res. Commun. 2017, 482, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Kiriyama, Y.; Nochi, H. Intra- and Intercellular Quality Control Mechanisms of Mitochondria. Cells 2017, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Russell, O.M.; Gorman, G.S.; Lightowlers, R.N.; Turnbull, D.M. Mitochondrial Diseases: Hope for the Future. Cell 2020, 181, 168–188. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Ghochani, M.; McCaffery, J.M.; Frey, T.G.; Chan, D.C. Mitofusins and OPA1 Mediate Sequential Steps in Mitochondrial Membrane Fusion. Mol. Biol. Cell 2009, 20, 3525–3532. [Google Scholar] [CrossRef]
- Koshiba, T.; Detmer, S.A.; Kaiser, J.T.; Chen, H.; McCaffery, J.M.; Chan, D.C. Structural Basis of Mitochondrial Tethering by Mitofusin Complexes. Science 2004, 305, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Hu, J. Mitochondrial Fusion: The Machineries In and Out. Trends Cell Biol. 2020, 31, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhou, X.; Hu, X.; Joshi, A.S.; Guo, X.; Zhu, Y.; Chen, Q.; Prinz, W.A.; Hu, J. Sequences flanking the transmembrane segments facilitate mitochondrial localization and membrane fusion by mitofusin. Proc. Natl. Acad. Sci. USA 2017, 114, E9863–E9872. [Google Scholar] [CrossRef]
- Ishihara, N.; Eura, Y.; Mihara, K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J. Cell Sci. 2004, 117, 6535–6546. [Google Scholar] [CrossRef]
- Han, S.; Zhao, F.; Hsia, J.; Ma, X.; Liu, Y.; Torres, S.; Fujioka, H.; Zhu, X. The role of Mfn2 in the structure and function of endoplasmic reticulum-mitochondrial tethering in vivo. J. Cell Sci. 2021, 134, jcs253443. [Google Scholar] [CrossRef] [PubMed]
- Zaman, M.; Shutt, T.E. The Role of Impaired Mitochondrial Dynamics in MFN2-Mediated Pathology. Front. Cell Dev. Biol. 2022, 10, 858286. [Google Scholar] [CrossRef] [PubMed]
- Kraus, F.; Roy, K.; Pucadyil, T.J.; Ryan, M.T. Function and regulation of the divisome for mitochondrial fission. Nature 2021, 590, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Zhou, Y.; Lu, Y.; Zhou, K.; Cai, W. PHB2 interacts with LC3 and SQSTM1 is required for bile acids-induced mitophagy in cholestatic liver. Cell Death Dis. 2018, 9, 160. [Google Scholar] [CrossRef]
- Palmeira, C.M.; Rolo, A.P. Mitochondrially-mediated toxicity of bile acids. Toxicology 2004, 203, 1–15. [Google Scholar] [CrossRef]
- Perez, M.-J.; Briz, O. Bile-acid-induced cell injury and protection. World J. Gastroenterol. 2009, 15, 1677–1689. [Google Scholar] [CrossRef] [PubMed]
- Popov, L. Mitochondrial biogenesis: An update. J. Cell. Mol. Med. 2020, 24, 4892–4899. [Google Scholar] [CrossRef]
- Zhao, L.-J.; Zhang, S.-F. Activation of TGR5 promotes mitochondrial biogenesis in human aortic endothelial cells. Biochem. Biophys. Res. Commun. 2018, 500, 952–957. [Google Scholar] [CrossRef]
- Liu, J.; Yang, J. Mitochondria-associated membranes: A hub for neurodegenerative diseases. Biomed. Pharmacother. 2022, 149, 112890. [Google Scholar] [CrossRef]
- Markovinovic, A.; Greig, J.; Martín-Guerrero, S.M.; Salam, S.; Paillusson, S. Endoplasmic reticulum–mitochondria signaling in neurons and neurodegenerative diseases. J. Cell Sci. 2022, 135, jcs248534. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER–mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef]
- Gomez-Suaga, P.; Paillusson, S.; Miller, C.C.J. ER-mitochondria signaling regulates autophagy. Autophagy 2017, 13, 1250–1251. [Google Scholar] [CrossRef]
- Ding, Y.; Gao, H.; Zhao, L.; Wang, X.; Zheng, M. Mitofusin 2-Deficiency Suppresses Cell Proliferation through Disturbance of Autophagy. PLoS ONE 2015, 10, e0121328. [Google Scholar] [CrossRef]
- Ashraf, R.; Kumar, S. Mfn2-mediated mitochondrial fusion promotes autophagy and suppresses ovarian cancer progression by reducing ROS through AMPK/mTOR/ERK signaling. Cell. Mol. Life Sci. 2022, 79, 573. [Google Scholar] [CrossRef]
- Filadi, R.; Pendin, D.; Pizzo, P. Mitofusin 2: From functions to disease. Cell Death Dis. 2018, 9, 330. [Google Scholar] [CrossRef]
- Patra, S.; Patil, S.; Klionsky, D.J.; Bhutia, S.K. Lysosome signaling in cell survival and programmed cell death for cellular homeostasis. J. Cell. Physiol. 2022, 238, 287–305. [Google Scholar] [CrossRef] [PubMed]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in healthy aging and disease. Nat. Aging 2021, 1, 634–650. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The Hairpin-type Tail-Anchored SNARE Syntaxin 17 Targets to Autophagosomes for Fusion with Endosomes/Lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [PubMed]
- Atlashkin, V.; Kreykenbohm, V.; Eskelinen, E.-L.; Wenzel, D.; Fayyazi, A.; von Mollard, G.F. Deletion of the SNARE vti1b in Mice Results in the Loss of a Single SNARE Partner, Syntaxin 8. Mol. Cell. Biol. 2003, 23, 5198–5207. [Google Scholar] [CrossRef]
- Gutierrez, M.G.; Munafó, D.B.; Berón, W.; Colombo, M.I. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J. Cell Sci. 2004, 117, 2687–2697. [Google Scholar] [CrossRef] [PubMed]
- Jäger, S.; Bucci, C.; Tanida, I.; Ueno, T.; Kominami, E.; Saftig, P.; Eskelinen, E.-L. Role for Rab7 in maturation of late autophagic vacuoles. J. Cell Sci. 2004, 117, 4837–4848. [Google Scholar] [CrossRef]
- Liang, C.; Lee, J.-S.; Inn, K.-S.; Gack, M.U.; Li, Q.; Roberts, E.A.; Vergne, I.; Deretic, V.; Feng, P.; Akazawa, C.; et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nature 2008, 10, 776–787. [Google Scholar] [CrossRef]
- Jiang, P.; Nishimura, T.; Sakamaki, Y.; Itakura, E.; Hatta, T.; Natsume, T.; Mizushima, N. The HOPS complex mediates autophagosome–lysosome fusion through interaction with syntaxin 17. Mol. Biol. Cell 2014, 25, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, D.S.; Jariwala, J.S.; Anderson, E.; Mitra, K.; Meisenhelder, J.; Chang, J.T.; Ideker, T.; Hunter, T.; Nizet, V.; Dillin, A.; et al. Phosphorylation of LC3 by the Hippo Kinases STK3/STK4 Is Essential for Autophagy. Mol. Cell 2014, 57, 55–68. [Google Scholar] [CrossRef]
- Wang, H.; Sun, H.-Q.; Zhu, X.; Zhang, L.; Albanesi, J.; Levine, B.; Yin, H. GABARAPs regulate PI4P-dependent autophagosome:lysosome fusion. Proc. Natl. Acad. Sci. USA 2015, 112, 7015–7020. [Google Scholar] [CrossRef] [PubMed]
- Lan, W.; Chen, Z.; Chen, Y.; Tan, M.; Chen, Y.; Chen, J.; Chi, X.; Chen, Y. Glycochenodeoxycholic acid impairs transcription factor E3 -dependent autophagy-lysosome machinery by disrupting reactive oxygen species homeostasis in L02 cells. Toxicol. Lett. 2020, 331, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Seok, S.; Fu, T.; Choi, S.-E.; Li, Y.; Zhu, R.; Kumar, S.; Sun, X.; Yoon, G.; Kang, Y.; Zhong, W.; et al. Transcriptional regulation of autophagy by an FXR–CREB axis. Nature 2014, 516, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Wagner, M.; Xiao, R.; Kim, K.H.; Feng, D.; Lazar, M.A.; Moore, D.D. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 2014, 516, 112–115. [Google Scholar] [CrossRef]
- Carino, A.; Marchianò, S.; Biagioli, M.; Scarpelli, P.; Bordoni, M.; Di Giorgio, C.; Roselli, R.; Fiorucci, C.; Monti, M.C.; Distrutti, E.; et al. The bile acid activated receptors GPBAR1 and FXR exert antagonistic effects on autophagy. FASEB J. 2020, 35, e21271. [Google Scholar] [CrossRef]
- Pan, Y.; He, X.; Li, C.; Li, Y.; Li, W.; Zhang, H.; Wang, Y.; Zhou, G.; Yang, J.; Li, J.; et al. Neuronal activity recruits the CRTC1/CREB axis to drive transcription-dependent autophagy for maintaining late-phase LTD. Cell Rep. 2021, 36, 109398. [Google Scholar] [CrossRef] [PubMed]
- Baloni, P.; Funk, C.C.; Yan, J.; Yurkovich, J.T.; Kueider-Paisley, A.; Nho, K.; Heinken, A.; Jia, W.; Mahmoudiandehkordi, S.; Louie, G.; et al. Metabolic Network Analysis Reveals Altered Bile Acid Synthesis and Metabolism in Alzheimer’s Disease. Cell Rep. Med. 2020, 1, 100138. [Google Scholar] [CrossRef]
- Mano, N.; Goto, T.; Uchida, M.; Nishimura, K.; Ando, M.; Kobayashi, N.; Goto, J. Presence of protein-bound unconjugated bile acids in the cytoplasmic fraction of rat brain. J. Lipid Res. 2004, 45, 295–300. [Google Scholar] [CrossRef]
- Pan, X.; Elliott, C.T.; McGuinness, B.; Passmore, P.; Kehoe, P.G.; Hölscher, C.; McClean, P.L.; Graham, S.F.; Green, B.D. Metabolomic Profiling of Bile Acids in Clinical and Experimental Samples of Alzheimer’s Disease. Metabolites 2017, 7, 28. [Google Scholar] [CrossRef]
- Mertens, K.L.; Kalsbeek, A.; Soeters, M.R.; Eggink, H.M. Bile Acid Signaling Pathways from the Enterohepatic Circulation to the Central Nervous System. Front. Neurosci. 2017, 11, 617. [Google Scholar] [CrossRef]
- Higashi, T.; Watanabe, S.; Tomaru, K.; Yamazaki, W.; Yoshizawa, K.; Ogawa, S.; Nagao, H.; Minato, K.; Maekawa, M.; Mano, N. Unconjugated bile acids in rat brain: Analytical method based on LC/ESI-MS/MS with chemical derivatization and estimation of their origin by comparison to serum levels. Steroids 2017, 125, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Kamp, F.; Hamilton, J.A. Movement of fatty acids, fatty acid analogs, and bile acids across phospholipid bilayers. Biochemistry 1993, 32, 11074–11085. [Google Scholar] [CrossRef] [PubMed]
- Monteiro-Cardoso, V.F.; Corlianò, M.; Singaraja, R.R. Bile Acids: A Communication Channel in the Gut-Brain Axis. NeuroMol. Med. 2020, 23, 99–117. [Google Scholar] [CrossRef]
- Benedetti, A.; Di Sario, A.; Marucci, L.; Svegliati-Baroni, G.; Schteingart, C.D.; Ton-Nu, H.T.; Hofmann, A.F. Carrier-mediated transport of conjugated bile acids across the basolateral membrane of biliary epithelial cells. Am. J. Physiol. Liver Physiol. 1997, 272, G1416–G1424. [Google Scholar] [CrossRef]
- St-Pierre, M.V.; Kullak-Ublick, G.A.; Hagenbuch, B.; Meier, P.J. Transport of Bile Acids in Hepatic and Non-Hepatic Tissues. J. Exp. Biol. 2001, 204, 1673–1686. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s Disease: The Challenge of the Second Century. Sci. Transl. Med. 2011, 3, 77sr1. [Google Scholar] [CrossRef]
- Vassar, R.; Cole, S. The Basic Biology of BACE1: A Key Therapeutic Target for Alzheimers Disease. Curr. Genom. 2007, 8, 509–530. [Google Scholar] [CrossRef]
- Tomita, T. Molecular mechanism of intramembrane proteolysis by γ-secretase. J. Biochem. 2014, 156, 195–201. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, X.; Song, Y.-Q.; Tu, J. Autophagy in Alzheimer’s disease pathogenesis: Therapeutic potential and future perspectives. Ageing Res. Rev. 2021, 72, 101464. [Google Scholar] [CrossRef] [PubMed]
- Marksteiner, J.; Blasko, I.; Kemmler, G.; Koal, T.; Humpel, C. Bile acid quantification of 20 plasma metabolites identifies lithocholic acid as a putative biomarker in Alzheimer’s disease. Metabolomics 2017, 14, 1. [Google Scholar] [CrossRef]
- Mulak, A. Bile Acids as Key Modulators of the Brain-Gut-Microbiota Axis in Alzheimer’s Disease. J. Alzheimers Dis. 2021, 84, 461–477. [Google Scholar] [CrossRef]
- Nunes, A.F.; Amaral, J.D.; Lo, A.C.; Fonseca, M.B.; Viana, R.J.; Callaerts-Vegh, Z.; D’Hooge, R.; Rodrigues, C.M. TUDCA, a bile acid, attenuates amyloid precursor protein processing and amyloid-β deposition in APP/PS1 mice. Mol. Neurobiol. 2012, 45, 440–454. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.C.; Callaerts-Vegh, Z.; Nunes, A.F.; Rodrigues, C.M.; D’Hooge, R. Tauroursodeoxycholic acid (TUDCA) supplementation prevents cognitive impairment and amyloid deposition in APP/PS1 mice. Neurobiol. Dis. 2013, 50, 21–29. [Google Scholar] [CrossRef] [PubMed]
- MahmoudianDehkordi, S.; Arnold, M.; Nho, K.; Ahmad, S.; Jia, W.; Xie, G.; Louie, G.; Kueider-Paisley, A.; Moseley, M.A.; Thompson, J.W.; et al. Altered bile acid profile associates with cognitive impairment in Alzheimer’s disease—An emerging role for gut microbiome. Alzheimers Dement. 2019, 15, 76–92. [Google Scholar] [CrossRef]
- Wu, S.; Liu, X.; Jiang, R.; Yan, X.; Ling, Z. Roles and Mechanisms of Gut Microbiota in Patients With Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 650047. [Google Scholar] [CrossRef]
- D’Argenio, V.; Veneruso, I.; Gong, C.; Cecarini, V.; Bonfili, L.; Eleuteri, A.M. Gut Microbiome and Mycobiome Alterations in an In Vivo Model of Alzheimer’s Disease. Genes 2022, 13, 1564. [Google Scholar] [CrossRef]
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Brichta, L.; Greengard, P.; Flajolet, M. Advances in the pharmacological treatment of Parkinson’s disease: Targeting neurotransmitter systems. Trends Neurosci. 2013, 36, 543–554. [Google Scholar] [CrossRef]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Eldeeb, M.A.; Thomas, R.A.; Ragheb, M.A.; Fallahi, A.; Fon, E.A. Mitochondrial quality control in health and in Parkinson’s disease. Physiol. Rev. 2022, 102, 1721–1755. [Google Scholar] [CrossRef]
- Yakhine-Diop, S.M.; Morales-García, J.A.; Niso-Santano, M.; González-Polo, R.A.; Uribe-Carretero, E.; Martinez-Chacon, G.; Durand, S.; Maiuri, M.C.; Aiastui, A.; Zulaica, M.; et al. Metabolic alterations in plasma from patients with familial and idiopathic Parkinson’s disease. Aging 2020, 12, 16690–16708. [Google Scholar] [CrossRef]
- Shao, Y.; Li, T.; Liu, Z.; Wang, X.; Xu, X.; Li, S.; Xu, G.; Le, W. Comprehensive metabolic profiling of Parkinson’s disease by liquid chromatography-mass spectrometry. Mol. Neurodegener. 2021, 16, 4. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, C.; Zhao, N.; Li, W.; Yang, Z.; Liu, X.; Le, W.; Zhang, X. Potential biomarkers of Parkinson’s disease revealed by plasma metabolic profiling. J. Chromatogr. B 2018, 1081–1082, 101–108. [Google Scholar] [CrossRef]
- Li, P.; Killinger, B.; Ensink, E.; Beddows, I.; Yilmaz, A.; Lubben, N.; Lamp, J.; Schilthuis, M.; Vega, I.; Woltjer, R.; et al. Gut Microbiota Dysbiosis Is Associated with Elevated Bile Acids in Parkinson’s Disease. Metabolites 2021, 11, 29. [Google Scholar] [CrossRef]
- Martinez, T.N.; Greenamyre, J.T.; Ritz, B.R.; Paul, K.C.; Bronstein, J.M.; Lee, J.; Fifel, K.; Piggins, H.; Deboer, T.; Martella, G.; et al. Toxin Models of Mitochondrial Dysfunction in Parkinson’s Disease. Antioxid. Redox Signal. 2012, 16, 920–934. [Google Scholar] [CrossRef] [PubMed]
- Connolly, N.M.C.; Theurey, P.; Adam-Vizi, V.; Bazan, N.G.; Bernardi, P.; Bolaños, J.P.; Culmsee, C.; Dawson, V.L.; Deshmukh, M.; Duchen, M.R.; et al. Guidelines on experimental methods to assess mitochondrial dysfunction in cellular models of neurodegenerative diseases. Cell Death Differ. 2017, 25, 542–572. [Google Scholar] [CrossRef] [PubMed]
- Castro-Caldas, M.; Carvalho, A.N.; Rodrigues, E.; Henderson, C.; Wolf, C.R.; Rodrigues, C.; Gama, M.J. Tauroursodeoxycholic Acid Prevents MPTP-Induced Dopaminergic Cell Death in a Mouse Model of Parkinson’s Disease. Mol. Neurobiol. 2012, 46, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.I.; Duarte-Silva, S.; Silva-Fernandes, A.; Nunes, M.J.; Carvalho, A.N.; Rodrigues, E.; Gama, M.J.; Rodrigues, C.M.P.; Maciel, P.; Castro-Caldas, M. Tauroursodeoxycholic Acid Improves Motor Symptoms in a Mouse Model of Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 9139–9155. [Google Scholar] [CrossRef] [PubMed]
- Abdelkader, N.F.; Safar, M.M.; Salem, H.A. Ursodeoxycholic Acid Ameliorates Apoptotic Cascade in the Rotenone Model of Parkinson’s Disease: Modulation of Mitochondrial Perturbations. Mol. Neurobiol. 2014, 53, 810–817. [Google Scholar] [CrossRef]
- Dayalu, P.; Albin, R.L. Huntington Disease. Neurol. Clin. 2015, 33, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington Disease. Nat. Rev. Dis. Prim. 2015, 1, 15005. [Google Scholar] [CrossRef] [PubMed]
- Rui, Y.-N.; Xu, Z.; Patel, B.; Chen, Z.; Chen, D.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.J.; et al. Huntingtin functions as a scaffold for selective macroautophagy. Nature 2015, 17, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Steffan, J.S. Does Huntingtin play a role in selective macroautophagy? Cell Cycle 2010, 9, 3401–3413. [Google Scholar] [CrossRef] [PubMed]
- Wanker, E.E.; Ast, A.; Schindler, F.; Trepte, P.; Schnoegl, S. The pathobiology of perturbed mutant huntingtin protein–protein interactions in Huntington’s disease. J. Neurochem. 2019, 151, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Franco-Iborra, S.; Plaza-Zabala, A.; Montpeyo, M.; Sebastian, D.; Vila, M.; Martinez-Vicente, M. Mutant HTT (huntingtin) impairs mitophagy in a cellular model of Huntington disease. Autophagy 2020, 17, 672–689. [Google Scholar] [CrossRef] [PubMed]
- Túnez, I.; Tasset, I.; La Cruz, V.P.-D.; Santamaría, A. 3-Nitropropionic Acid as a Tool to Study the Mechanisms Involved in Huntington’s Disease: Past, Present and Future. Molecules 2010, 15, 878–916. [Google Scholar] [CrossRef] [PubMed]
- Brouillet, E.; Jacquard, C.; Bizat, N.; Blum, D. 3-Nitropropionic acid: A mitochondrial toxin to uncover physiopathological mechanisms underlying striatal degeneration in Huntington’s disease. J. Neurochem. 2005, 95, 1521–1540. [Google Scholar] [CrossRef] [PubMed]
- Keene, C.; Rodrigues, C.M.; Eich, T.; Linehan-Stieers, C.; Abt, A.; Kren, B.T.; Steer, C.J.; Low, W.C. A Bile Acid Protects against Motor and Cognitive Deficits and Reduces Striatal Degeneration in the 3-Nitropropionic Acid Model of Huntington’s Disease. Exp. Neurol. 2001, 171, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Keene, C.D.; Rodrigues, C.M.P.; Eich, T.; Chhabra, M.S.; Steer, C.J.; Low, W.C. Tauroursodeoxycholic acid, a bile acid, is neuroprotective in a transgenic animal model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2002, 99, 10671–10676. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiriyama, Y.; Nochi, H. Role of Microbiota-Modified Bile Acids in the Regulation of Intracellular Organelles and Neurodegenerative Diseases. Genes 2023, 14, 825. https://doi.org/10.3390/genes14040825
Kiriyama Y, Nochi H. Role of Microbiota-Modified Bile Acids in the Regulation of Intracellular Organelles and Neurodegenerative Diseases. Genes. 2023; 14(4):825. https://doi.org/10.3390/genes14040825
Chicago/Turabian StyleKiriyama, Yoshimitsu, and Hiromi Nochi. 2023. "Role of Microbiota-Modified Bile Acids in the Regulation of Intracellular Organelles and Neurodegenerative Diseases" Genes 14, no. 4: 825. https://doi.org/10.3390/genes14040825
APA StyleKiriyama, Y., & Nochi, H. (2023). Role of Microbiota-Modified Bile Acids in the Regulation of Intracellular Organelles and Neurodegenerative Diseases. Genes, 14(4), 825. https://doi.org/10.3390/genes14040825