
Rare 15q21.1q22.31 Duplication Due to a Familial Chromosomal Insertion and Diagnostic Investigation in a Carrier of Balanced Chromosomal Rearrangement and Intellectual Disability
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cytogenetic Analysis
2.2. Chromosomal Microarray Analysis (CMA)
2.3. Low-Pass Whole Genome Sequencing (WGS) and Whole Exome Sequencing (WES)
3. Results
3.1. Cases Presentation
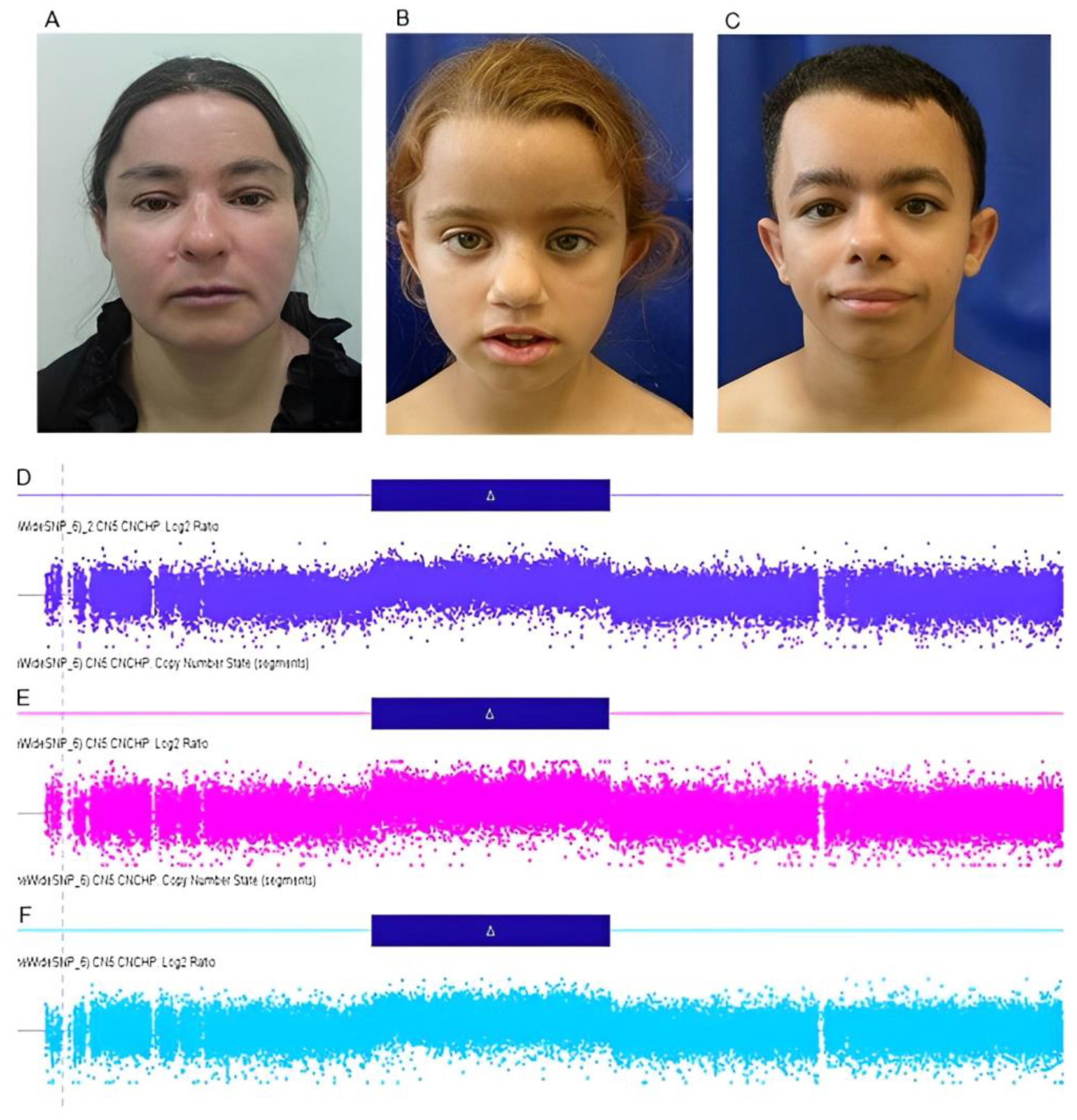
3.1.1. Subject 1 (III-4)
3.1.2. Subject 2 (IV-11)
3.1.3. Subject 3 (IV-12)
3.1.4. Subject 4 (IV-15)
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Hemel, J.O.; Eussen, H.J. Interchromosomal Insertions. Identification of Five Cases and a Review. Hum. Genet. 2000, 107, 415–432. [Google Scholar] [CrossRef]
- Neill, N.J.; Ballif, B.C.; Lamb, A.N.; Parikh, S.; Ravnan, J.B.; Schultz, R.A.; Torchia, B.S.; Rosenfeld, J.A.; Shaffer, L.G. Recurrence, Submicroscopic Complexity, and Potential Clinical Relevance of Copy Gains Detected by Array CGH That Are Shown to Be Unbalanced Insertions by FISH. Genome Res. 2011, 21, 535–544. [Google Scholar] [CrossRef] [Green Version]
- Nowakowska, B.A.; De Leeuw, N.; Ruivenkamp, C.A.L.; Sikkema-Raddatz, B.; Crolla, J.A.; Thoelen, R.; Koopmans, M.; Den Hollander, N.; Van Haeringen, A.; Van Der Kevie-Kersemaekers, A.M.; et al. Parental Insertional Balanced Translocations Are an Important Cause of Apparently de Novo CNVs in Patients with Developmental Anomalies. Eur. J. Hum. Genet. 2012, 20, 166–170. [Google Scholar] [CrossRef] [Green Version]
- Courtens, W.; Vroman, S.; Vandenhove, J.; Wiedemann, U.; Schinzel, A. Pre- and Perinatal Findings in Partial Trisomy 7q Resulting from Balanced Parental Translocations t(7;21) and t(4;7). Prenat. Diagn. 2001, 21, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Capra, V.; Mascelli, S.; Garrè, M.L.; Nozza, P.; Vaccari, C.; Bricco, L.; Sloan-Béna, F.; Gimelli, S.; Cuoco, C.; Gimelli, G.; et al. Parental Imbalances Involving Chromosomes 15q and 22q May Predispose to the Formation of de Novo Pathogenic Microdeletions and Microduplications in the Offspring. PLoS ONE 2013, 8, e57910. [Google Scholar] [CrossRef] [PubMed]
- Steiner, M.B.; Vengoechea, J.; Collins, R.T. Duplication of the ALDH1A2 Gene in Association with Pentalogy of Cantrell: A Case Report. J. Med. Case Rep. 2013, 7, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, X.; Wu, X.; Chen, J.; Wu, Y.; Li, S.; Chen, X.; Zhang, X. Aromatase Excess Syndrome in a Chinese Boy Due to a Novel Duplication at 15q21.2. J. Pediatr. Endocrinol. Metab. 2019, 32, 85–88. [Google Scholar] [CrossRef]
- Lindsay, F.; Anderson, I.; Wentzensen, I.M.; Suhrbier, D.; Stevens, C.A. Genetic Evaluation Including Exome Sequencing of Two Patients with Gomez-Lopez-Hernandez Syndrome: Case Reports and Review of the Literature. Am. J. Med. Genet. A 2020, 182, 623–627. [Google Scholar] [CrossRef]
- Chen, C.P.; Ko, T.M.; Wang, L.K.; Chern, S.R.; Wu, P.S.; Chen, S.W.; Wu, F.T.; Chen, L.F.; Wang, W. Prenatal Diagnosis of Partial Monosomy 8p (8p23.2→pter) and Partial Trisomy 15q (15q21.2→qter) and Incidental Detection of a Familial Chromosome Translocation of Paternal Origin in a Pregnancy Associated with Increased Nuchal Translucency and an Abnormal Maternal Serum Screening Result. Taiwan. J. Obstet. Gynecol. 2021, 60, 775–777. [Google Scholar] [CrossRef]
- Firth, H.V.; Richards, S.M.; Bevan, A.P.; Clayton, S.; Corpas, M.; Rajan, D.; Van Vooren, S.; Moreau, Y.; Pettett, R.M.; Carter, N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 2009, 84, 524–533. [Google Scholar] [CrossRef] [Green Version]
- Schluth-Bolard, C.; Labalme, A.; Cordier, M.P.; Till, M.; Nadeau, G.; Tevissen, H.; Lesca, G.; Boutry-Kryza, N.; Rossignol, S.; Rocas, D.; et al. Breakpoint Mapping by next Generation Sequencing Reveals Causative Gene Disruption in Patients Carrying Apparently Balanced Chromosome Rearrangements with Intellectual Deficiency and/or Congenital Malformations. J. Med. Genet. 2013, 50, 144–150. [Google Scholar] [CrossRef]
- Sismani, C.; Kitsiou-Tzeli, S.; Ioannides, M.; Christodoulou, C.; Anastasiadou, V.; Stylianidou, G.; Papadopoulou, E.; Kanavakis, E.; Kosmaidou-Aravidou, Z.; Patsalis, P.C. Cryptic Genomic Imbalances in Patients with de Novo or Familial Apparently Balanced Translocations and Abnormal Phenotype. Mol. Cytogenet. 2008, 1, 15. [Google Scholar] [CrossRef] [Green Version]
- Simioni, M.; Artiguenave, F.; Meyer, V.; Sgardioli, I.C.; Viguetti-Campos, N.L.; Lopes Monlleó, I.; MacIel-Guerra, A.T.; Steiner, C.E.; Gil-Da-Silva-Lopes, V.L. Genomic Investigation of Balanced Chromosomal Rearrangements in Patients with Abnormal Phenotypes. Mol. Syndromol. 2017, 8, 187–194. [Google Scholar] [CrossRef] [Green Version]
- Lelieveld, S.H.; Reijnders, M.R.F.; Pfundt, R.; Yntema, H.G.; Kamsteeg, E.J.; De Vries, P.; De Vries, B.B.A.; Willemsen, M.H.; Kleefstra, T.; Löhner, K.; et al. Meta-Analysis of 2,104 Trios Provides Support for 10 New Genes for Intellectual Disability. Nat. Neurosci. 2016, 19, 1194–1196. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Arens, Y.H.J.M.; Engelen, J.J.M.; Govaerts, L.C.P.; Van Ravenswaay, C.M.; Loneus, W.H.; Van Lent-Albrechts, J.C.M.; Van Der Blij-Philipsen, M.; Hamers, A.J.H.; Schrander-Stumpel, C.T.R.M. Familial Insertion (3;5)(Q25.3;Q22.1q31.3) with Deletion or Duplication of Chromosome Region 5q22.1-5q31.3 in Ten Unbalanced Carriers. Am. J. Med. Genet. A 2004, 130A, 128–133. [Google Scholar] [CrossRef]
- Fogu, G.; Bandiera, P.; Cambosu, F.; Carta, A.R.; Pilo, L.; Serra, G.; Soro, G.; Tondi, M.; Tusacciu, G.; Montella, A. Pure Partial Trisomy of 6p12.1-P22.1 Secondary to a Familial 12/6 Insertion in Two Malformed Babies. Eur. J. Med. Genet. 2007, 50, 103–111. [Google Scholar] [CrossRef]
- Rodríguez, L.; Niebuhr, E.; García, A.; Martínez-Fernández, M.L.; Peña Segura, J.L. Be Careful with Familial Unbalanced Chromosome Abnormalities! Am. J. Med Genet. Part A 2008, 146A, 2005–2007. [Google Scholar] [CrossRef] [PubMed]
- Spreiz, A.; Müller, D.; Zotter, S.; Albrecht, U.; Baumann, M.; Fauth, C.; Erdel, M.; Zschocke, J.; Utermann, G.; Kotzot, D. Phenotypic Variability of a Deletion and Duplication 6q16.1 → Q21 Due to a Paternal Balanced Ins(7;6)(P15;Q16.1q21). Am. J. Med Genet. Part A 2010, 152A, 2762–2767. [Google Scholar] [CrossRef]
- Fernández, R.M.; Sánchez, J.; García-Díaz, L.; Peláez-Nora, Y.; González-Meneses, A.; Antiñolo, G.; Borrego, S. Interstitial 10p Deletion Derived from a Maternal Ins(16;10)(Q22;P13p15.2): Report of the First Familial Case of 10p Monosomy Affecting to Two Familial Members of Different Generations. Am. J. Med. Genet. A 2016, 170A, 1268–1273. [Google Scholar] [CrossRef] [PubMed]
- Collinson, M.N.; Roberts, S.E.; Crolla, J.A.; Dennis, N.R. A Familial Balanced Inverted Insertion Ins(15)(q15q13q11.2) Producing Prader-Willi Syndrome, Angelman Syndrome and Duplication of 15q11.2-Q13 in a Single Family: Importance of Differentiation from a Paracentric Inversion. Am. J. Med. Genet. A 2004, 126A, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Y.; Kim, K.H.; Jun, H.J.; Je, G.H.; Glotzbach, C.D.; Shaffer, L.G. Partial Trisomy of Chromosome 10(q22-q24) Due to Maternal Insertional Translocation (15;10). Am. J. Med. Genet. A 2004, 131, 190–193. [Google Scholar] [CrossRef]
- Simioni, M.; Vieira, T.P.; Sgardioli, I.C.; Freitas, É.L.; Rosenberg, C.; Maurer-Morelli, C.V.; Lopes-Cendes, I.; Fett-Conte, A.C.; Gil-da-Silva-Lopes, V.L. Insertional Translocation of 15q25-q26 into 11p13 and Duplication at 8p23.1 Characterized by High Resolution Arrays in a Boy with Congenital Malformations and Aniridia. Am. J. Med. Genet. A 2012, 158A, 2905–2910. [Google Scholar] [CrossRef] [PubMed]
- Türedi, O.; Vlcdan, A.; Yürürkutlay, N. Distal Trisomy 10q Due to Maternal Insertional Translocation (15;10): A Case Report and Review of Literature. Genet. Couns. 2016, 27, 273–278. [Google Scholar]
- Bailey, J.A.; Eichler, E.E. Genome-Wide Detection and Analysis of Recent Segmental Duplications within Mammalian Organisms. Cold Spring Harb. Symp. Quant. Biol. 2003, 68, 115–124. [Google Scholar] [CrossRef]
- Demura, M.; Martin, R.M.; Shozu, M.; Sebastian, S.; Takayama, K.; Hsu, W.-T.; Schultz, R.A.; Neely, K.; Bryant, M.; Mendonca, B.B.; et al. Regional Rearrangements in Chromosome 15q21 Cause Formation of Cryptic Promoters for the CYP19 (Aromatase) Gene. Hum. Mol. Genet. 2007, 16, 2529–2541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stratakis, C.A.; Vottero, A.; Brodie, A.; Kirschner, L.S.; Deatkine, D.; Lu, Q.; Yue, W.; Mitsiades, C.S.; Flor, A.W.; Chrousos, G.P. The Aromatase Excess Syndrome Is Associated with Feminization of Both Sexes and Autosomal Dominant Transmission of Aberrant P450 Aromatase Gene Transcription. J. Clin. Endocrinol. Metab. 1998, 83, 1348–1357. [Google Scholar] [CrossRef] [Green Version]
- Martin, R.M.; Lin, C.J.; Nishi, M.Y.; Billerbeck, A.E.C.; Latronico, A.C.; Russell, D.W.; Mendonca, B.B. Familial Hyperestrogenism in Both Sexes: Clinical, Hormonal, and Molecular Studies of Two Siblings. J. Clin. Endocrinol. Metab. 2003, 88, 3027–3034. [Google Scholar] [CrossRef] [Green Version]
- Oh, R.Y.; Deshwar, A.R.; Marwaha, A.; Sabha, N.; Tropak, M.; Hou, H.; Yuki, K.E.; Wilson, M.D.; Rump, P.; Lunsing, R.; et al. Biallelic Loss-of-Function Variants in RABGAP1 Cause a Novel Neurodevelopmental Syndrome. Genet. Med. 2022, 24, 2399–2407. [Google Scholar] [CrossRef]
- Aristidou, C.; Koufaris, C.; Theodosiou, A.; Bak, M.; Mehrjouy, M.M.; Behjati, F.; Tanteles, G.; Christophidou-Anastasiadou, V.; Tommerup, N.; Sismani, C. Accurate Breakpoint Mapping in Apparently Balanced Translocation Families with Discordant Phenotypes Using Whole Genome Mate-Pair Sequencing. PLoS ONE 2017, 12, e169935. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Present Case Reports | Cases From Literature | Cases From DECIPHER | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Subject 1 | Subject 2 | Subject 4 | [7] | [5] | [9] | [6] | [8] | 260222 | 395132 | 427560 | 251433 | 308734 | 317274 | 401711 | Total | |
| Gender | female | female | male | male | famale | male | male | male | male | male | male | female | female | male | female | |
| Chromosome region | 15q21.1q22.31 | 15q21.1q22.31 | 15q21.1q22.31 | 15q21.2 | 15q21.2q22.2 | 15q21.2q26.3 | 15q21.3 | 15q21.3 | 15q15.1q22.2 | 15p21.1q22.1 | 15q21.3q22.31 | 15q21.2q24.1 | 15q21.1q22.2 | 15q21.2q22.31 | 15q15.2q21.2 | |
| Genomic positions | chr15:46,487,891–65,795,296 | chr15:46,487,891–65,795,296 | chr15:46,487,891–65,795,296 | chr15:50,382,769–51,568,204 | chr15:51,604,508–62,102,756 | chr15:50,903,432–102,338,129 | chr15:57,529,846–58,949,448 | chr15:54,776,491–55,822,045 | chr15:40,848,086–60,105,306 | chr15:44,520,510–58,920,509 | chr15:57,052,168–66,127,560 | chr15:51,314,381–73,572,490 | chr15:46,072,402–61,905,184 | Chr15:50,554,375–63,890,439 | chr15:43,032,935–51,179,063 | |
| Duplication size (Mb) | 19.3 | 19.3 | 19.3 | 1.1 | 10.5 | 2.4 | 1.4 | 1.0 | 19.3 | 14.4 | 9.1 | 22.3 | 15.8 | 13.3 | 8.2 | |
| Intellectual disability | + | + | + | NR | − | NR | NR | − | + | NR | + | − | + | + | + | 08/15 |
| Neurodevelopmental delay | + | + | + | NR | + | NR | NR | + | NR | NR | + | − | − | + | − | 07/15 |
| Delayed speech and language development | + | + | + | NR | − | NR | NR | + | + | NR | + | + | − | + | − | 08/15 |
| Abnormal heart morphology | + | + | + | NR | − | NR | + | + | + | NR | NR | + | + | NR | NR | 08/15 |
| Short stature | + | + | + | + | NR | NR | NR | − | + | + | − | + | + | NR | + | 09/15 |
| Cubitus valgus | + | + | + | + | NR | NR | − | − | NR | NR | NR | NR | NR | NR | NR | 04/15 |
| Abnormal facial shape | + | + | + | NR | − | + | − | − | NR | + | − | − | NR | + | + | 07/15 |
| Strabismus | − | − | + | NR | − | NR | − | + | NR | + | − | − | NR | − | − | 03/15 |
| Low-set ears | + | + | + | NR | − | + | − | − | NR | + | + | + | NR | − | − | 07/15 |
| Accelerated skeletal maturation | + | + | + | + | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | 04/15 |
| Other features | Premature ovarian insufficiency. | Secondary amenorrhea. | Abnormal skeletal muscle morphology; Skeletal muscle hypertrophy; Velopharyngeal insufficiency; Hypothyroidism. | Gynecomastia. | Vascular malformation in the left maxillary sinus; | Fetal omphalocele; Tetralogy of Fallot. | Anxiety; Obsessive compulsive tendencies; Inability to walk; Motor stereotypy. | Micropenis; Delayed cranial suture closure; Seizures; Delayed skeletal maturation. | Cryptorchidism; Hypospadias; Small scrotum; Obesity. | Hypotonia. | Ptosis. | Early onset of sexual maturation. | Macrotia; Hypertelorism; Proptosis; Pes planus; Delayed skeletal maturation. | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nascimento, C.G.; Prota, J.R.M.; Sgardioli, I.C.; Spineli-Silva, S.; Campos, N.L.V.; Gil-da-Silva-Lopes, V.L.; Vieira, T.P. Rare 15q21.1q22.31 Duplication Due to a Familial Chromosomal Insertion and Diagnostic Investigation in a Carrier of Balanced Chromosomal Rearrangement and Intellectual Disability. Genes 2023, 14, 885. https://doi.org/10.3390/genes14040885
Nascimento CG, Prota JRM, Sgardioli IC, Spineli-Silva S, Campos NLV, Gil-da-Silva-Lopes VL, Vieira TP. Rare 15q21.1q22.31 Duplication Due to a Familial Chromosomal Insertion and Diagnostic Investigation in a Carrier of Balanced Chromosomal Rearrangement and Intellectual Disability. Genes. 2023; 14(4):885. https://doi.org/10.3390/genes14040885
Chicago/Turabian StyleNascimento, Carolina Gama, Joana Rosa Marques Prota, Ilária Cristina Sgardioli, Samira Spineli-Silva, Nilma Lúcia Viguetti Campos, Vera Lúcia Gil-da-Silva-Lopes, and Társis Paiva Vieira. 2023. "Rare 15q21.1q22.31 Duplication Due to a Familial Chromosomal Insertion and Diagnostic Investigation in a Carrier of Balanced Chromosomal Rearrangement and Intellectual Disability" Genes 14, no. 4: 885. https://doi.org/10.3390/genes14040885