
The Complete Mitochondrial Genome of Mytilisepta virgata (Mollusca: Bivalvia), Novel Gene Rearrangements, and the Phylogenetic Relationships of Mytilidae
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection of Samples and DNA Extraction
2.2. Sequencing, Assembly, and Annotation of Mitochondrial Genomes
2.3. Visualization and Comparative Analysis of the Genome
2.4. Phylogenetic Analysis and Gene Order
2.5. Estimation of Divergence Times
3. Results
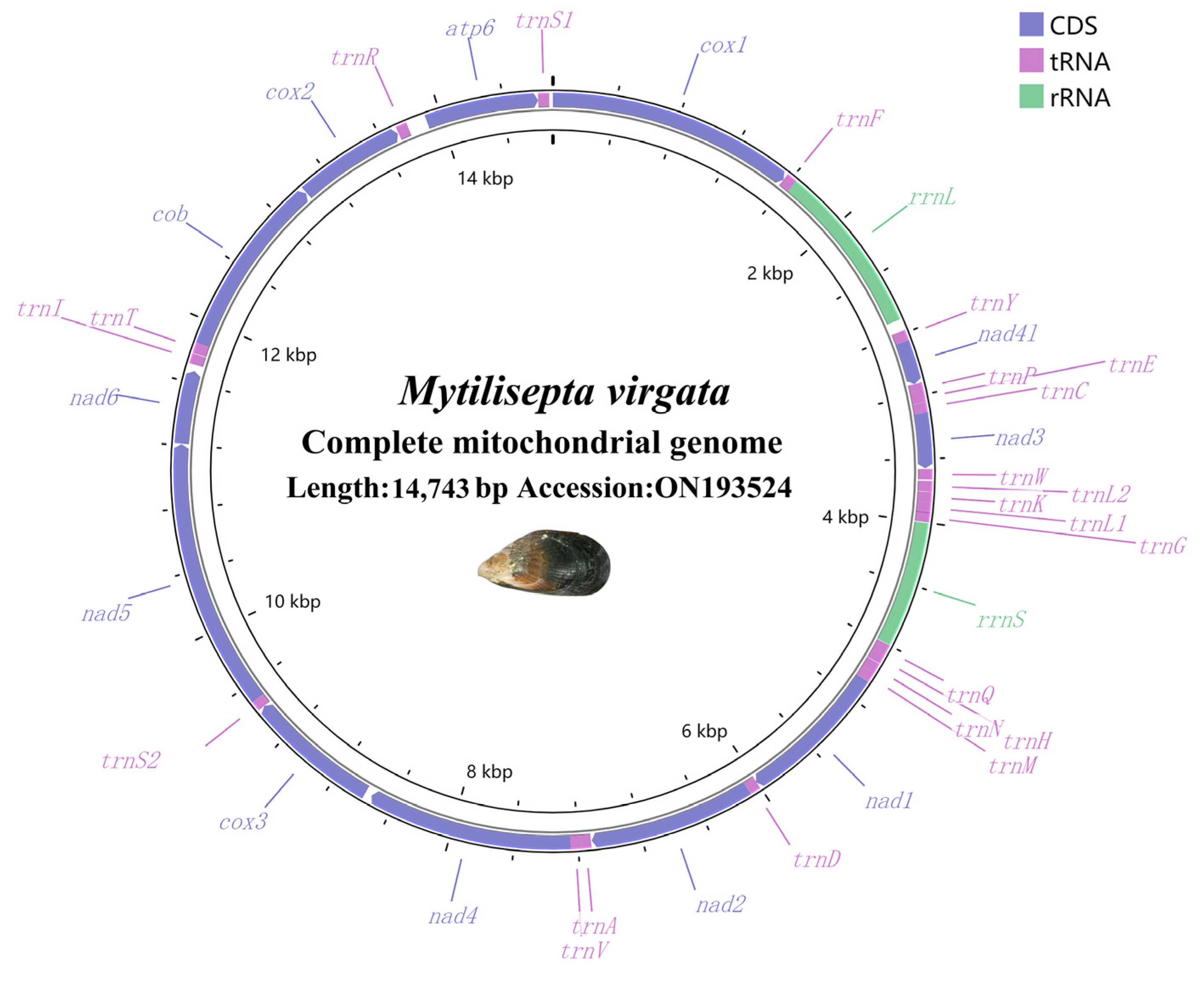
3.1. The Organization and Base Composition of the Genome
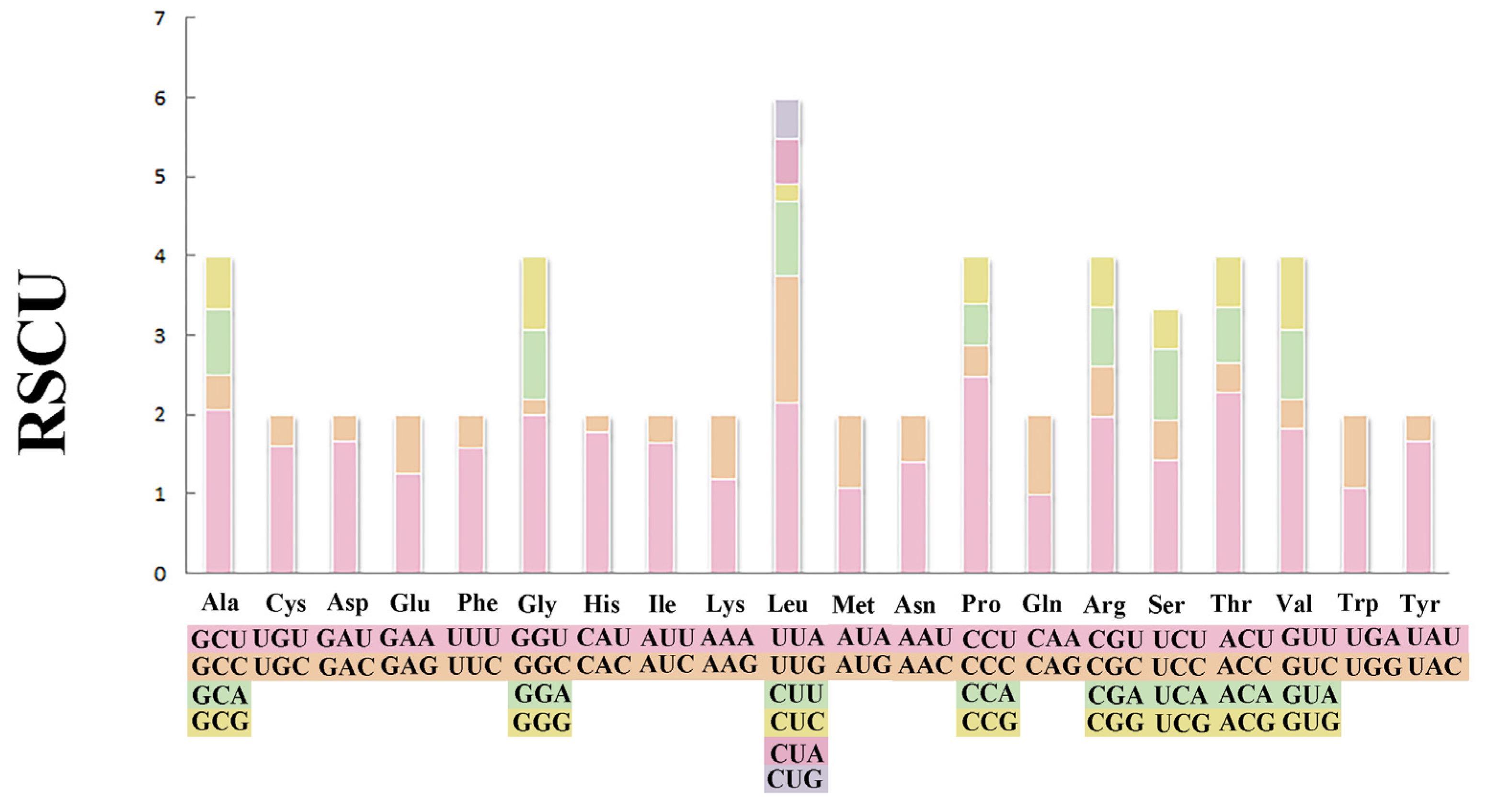
3.2. Use of Protein-Coding Genes and Codons
3.3. Transfer and Ribosomal RNA Genes
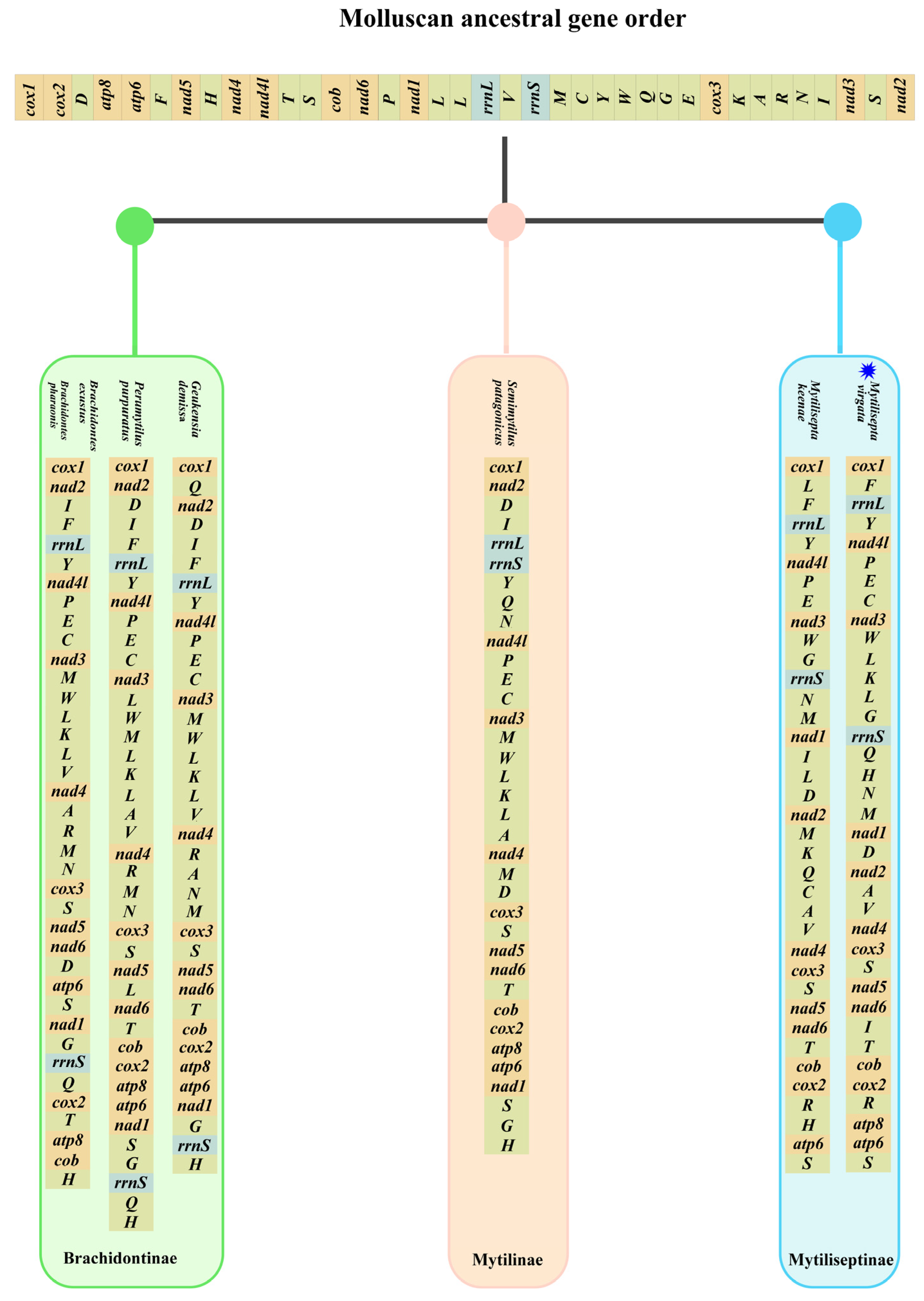
3.4. Gene Arrangement
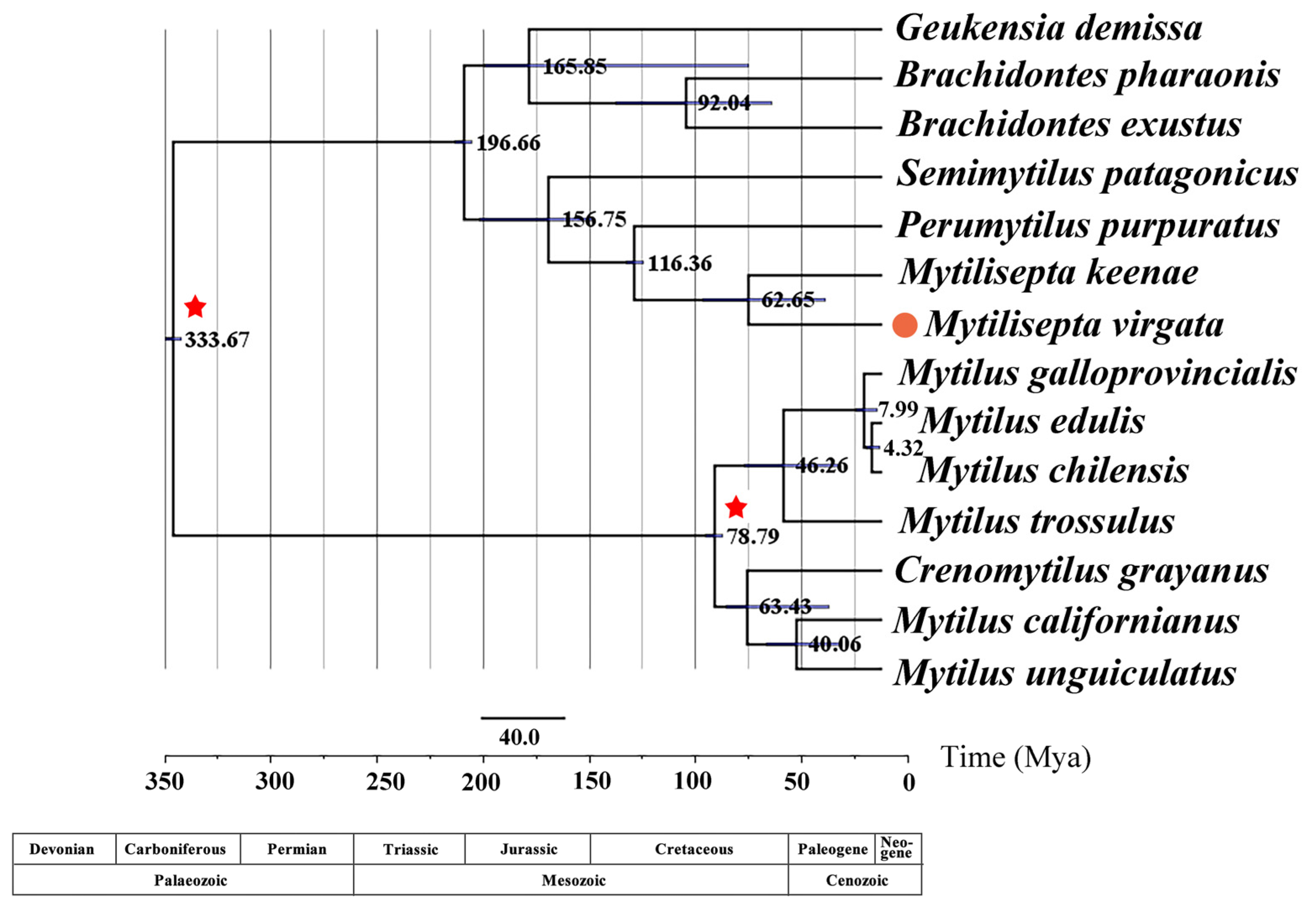
3.5. Phylogenetic Relationships of Mytilidae
3.6. Divergence Times
4. Discussion
4.1. The Organization of the Mitogenome
4.2. Protein Coding Genes and Codon Usage
4.3. tRNAs
4.4. Gene Arrangement
4.5. Phylogeny
4.6. Divergence Times
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gissi, C.; Iannelli, F.; Pesole, G. Evolution of the mitochondrial genome of metazoa as exemplified by comparison of congeneric species. Heredity 2008, 101, 301–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, G.L.; Köhler, F.; Huang, X.C.; Wu, R.W.; Zhou, C.H.; Ouyang, S.; Wu, X.P. A novel gene arrangement among the stylommatophora by the complete mitochondrial genome of the terrestrial slug Meghimatium bilineatum (Gastropoda, Arionoidea). Mol. Phylogenet. Evol. 2019, 135, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kwak, H.; Shin, J.; Kim, S.C.; Kim, T.; Park, J.K. A mitochondrial genome phylogeny of Mytilidae (Bivalvia: Mytilida). Mol. Phylogenet. Evol. 2019, 139, 106533. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kurokawa, T.; Sekino, M.; Tanabe, T.; Watanabe, K. Complete mitochondrial DNA sequence of the ark shell Scapharca broughtonii: An ultra-large metazoan mitochondrial genome. C.B.P.D Genom. Proteom. 2013, 8, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L.; Macey, J.R.; Medina, M. Sequencing and comparing whole mitochondrial genomes of animals. Methods Enzymol. 2005, 395, 311–348. [Google Scholar]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gérard, K.; Bierne, N.; Borsa, P.; Chenuil, A.; Féral, J. Pleistocene separation of mitochondrial lineages of Mytilus Spp. mussels from northern and southern hemispheres and strong genetic differentiation among southern populations. Mol. Phylogenet. Evol. 2008, 49, 84–91. [Google Scholar] [CrossRef]
- Krampah, E.A.; Yankson, K.; Blay, J. Population dynamics of the brown mussel Perna perna at a rocky beach near cape coast, ghana. Mar. Ecol. 2020, 41, e12571–e12575. [Google Scholar] [CrossRef]
- Colgan, D.J.; Costa, P.D. Invasive and non-invasive lineages in Xenostrobus (Bivalvia: Mytilidae). Molluscan Res. 2013, 33, 272–280. [Google Scholar] [CrossRef]
- Morton, B.; Dinesen, G.E. The biology and functional morphology of Modiolarca subpicta (Bivalvia: Mytilidae: Musculinae), epizoically symbiotic with Ascidiella aspersa (Urochordata: Ascidiacea), from the kattegat, northern jutland, denmark. J. Mar. Biol. Assoc. Uk 2011, 91, 1637–1649. [Google Scholar] [CrossRef]
- Lubośny, M.; Śmietanka, B.; Arculeo, M.; Burzyński, A. No evidence of DUI in the Mediterranean alien species Brachidontes pharaonis (P. Fisher, 1870) despite mitochondrial heteroplasmy. Sci. Rep. 2022, 12, 8569. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, K. Distribution and bed structure of the two intertidal mussels, Septifer virgatus (Wiegmann) and Hormomya mutabilis (Gould). P-SMBL 1994, 36, 223–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morton, B. The population dynamics and reproductive cycle of Septifer virgatus (Bivalvia: Mytilidae) on an exposed rocky shore in Hong Kong. J. Zool. 1995, 235, 485–500. [Google Scholar] [CrossRef]
- Okutani, T. Marine Mollusks in Japan; Tokai University Press: Tokai, Japan, 2000; 1173p. [Google Scholar]
- Liu, J.H.; Morton, B. The temperature tolerances of Tetraclita squamosa (Crustacea: Cirripedia) and Septifer virgatus (Bivalvia: Mytilidae) on a sub-tropical rocky shore in Hong Kong. J. Zool. 2010, 234, 325–339. [Google Scholar] [CrossRef]
- Seed, R. Patterns of biodiversity in the macro-invertebrate fauna associated with mussel patches on rocky shores. J. Mar. Biol. Assoc. Uk 1996, 76, 203–210. [Google Scholar] [CrossRef] [Green Version]
- Seed, R.; Richardson, C.A. Evolutionary traits in Perna viridis (Linnaeus) and Septifer virgatus (Wiegmann) (Bivalvia: Mytilidae). J. Exp. Mar. Biol. Ecol. 1999, 239, 273–287. [Google Scholar] [CrossRef]
- Gerdo, M.; Fujii, Y.; Hasan, I.; Koike, T.; Shimojo, S.; Spazzali, F.; Yamamoto, K.; Ozeki, Y.; Pallavicini, A.; Fujita, H. The purplish bifurcate mussel Mytilisepta virgata gene expression atlas reveals a remarkable tissue functional specialization. BMC Genom. 2017, 18, 590. [Google Scholar] [CrossRef] [Green Version]
- Yonge, C.M.; Campbell, J.I. II.-on the heteromyarian condition in the Bivalvia with special reference to dreissena polymorpha and certain Mytilacea. Earth Environ. Sci. Trans. R. Soc. Edinb. 1968, 68, 21–42. [Google Scholar] [CrossRef]
- Aljanabi, S.M.; Martinez, I. Universal and rapid salt-extraction of high quality genomic dna for Pcr-based techniques. Narnia 1997, 25, 4692–4693. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. Novoplasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. Mitos: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.R.; Stothard, P. The CGview server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. Mega X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. Trnascan-Se: A program for improved detection of transfer rna genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Laslett, D.; Canbck, A.B. Arwen: A program to detect trna genes in metazoan mitochondrial nucleotide sequences. BMC Bioinform. 2008, 24, 5–172. [Google Scholar] [CrossRef] [Green Version]
- Xia, X.; Zheng, X.; Salemi, M.; Lu, C.; Wang, Y. An index of substitution saturation and its application. Mol. Phylogenet. Evol. 2003, 26, 1–7. [Google Scholar] [CrossRef]
- Xia, X.; Xie, Z. Dambe: Software package for data analysis in molecular biology and evolution. J. Hered. 2001, 32, 371–373. [Google Scholar] [CrossRef] [Green Version]
- Lam-Tung, N.; Schmidt, H.A.; Arndt, V.H.; Quang, M.B. Iq-Tree: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Phylogenet. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.; Haeseler, A.V.; Jermiin, L.S. Modelfinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huelsenbeck, J.P. Mrbayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Swofford, D. Paup*: Phylogenetic analysis using parsimony (*and other methods). Evolution 2002, 56, 1776–1788. [Google Scholar] [CrossRef]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with beauti and the beast 1.7. Mol. Biol. Evol. 2012, 22, 1185–1192. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in bayesian phylogenetics using Tracer 1.7. Syst Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Uliano-Silva, M.; Americo, J.A.; Costa, I.; Schomaker-Bastos, A.; de Freitas Rebelo, M.; Prosdocimi, F. The complete mitochondrial genome of the golden mussel Limnoperna fortunei and comparative mitogenomics of mytilidae. Gene 2016, 577, 202–208. [Google Scholar] [CrossRef]
- Breton, S.; Stewart, D.T.; Hoeh, W.R. Characterization of a mitochondrial orf from the gender-associated mtdnas of Mytilus Spp. (Bivalvia: Mytilidae): Identification of the “missing” atpase 8 gene. Mar. Genom. 2010, 3, 11–18. [Google Scholar] [CrossRef]
- Fontanilla, I.K.; Naggs, F.; Wade, C.M. Molecular phylogeny of the Achatinoidea (Mollusca: Gastropoda). Mol. Phylogenet. Vol. 2017, 49, 114. [Google Scholar] [CrossRef]
- Serb, J.M.; Charles, L. Complete mtdna sequence of the north American freshwater mussel, Lampsilis ornata (Unionidae): An examination of the evolution and phylogenetic utility of mitochondrial genome organization in bivalvia (Mollusca). Mol. Biol. Evol. 2003, 20, 1854–1866. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yang, Y.; Liu, H.; Kong, L.; Li, Q. Phylogeny of Veneridae (Bivalvia) based on mitochondrial genomes. Zool. Scr. 2020, 50, 58–70. [Google Scholar] [CrossRef]
- Smith, D.R.; Snyder, M. Complete mitochondrial dna sequence of the scallop Placopecten magellanicus: Evidence of transposition leading to an uncharacteristically large mitochondrial genome. J. Mol. Evol. 2007, 65, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, X.; Li, L.; Xu, X.; Xia, J.; Yu, Z. New features of Asian Crassostrea oyster mitochondrial genomes: A novel alloacceptor trna gene recruitment and two novel orfs. Gene 2012, 507, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, R.J.; Boore, J.L.; Brown, W.M. A novel mitochondrial genome organization for the blue mussel, Mytilus edulis. Genetics 1992, 131, 397–412. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Sun, H.; Li, C.; Zhao, W.; Yao, Y. The complete mitochondrial genome of Platygaster robiniae (Hymenoptera: Platygastridae): A novel trna secondary structure, gene rearrangements and phylogenetic implications. Int. J. Parasitol. Par. 2022, 18, 249–259. [Google Scholar] [CrossRef]
- Gu, Y.L.; Sun, C.H.; Liu, P.; Zhang, X.; Sinev, A.Y.; Dumont, H.J.; Han, B.P. Complete mitochondrial genome of Ovalona pulchella (Branchiopoda, Cladocera) as the first representative in the family chydoridae: Gene rearrangements and phylogenetic analysis of cladocera. Gene 2022, 818, 146230. [Google Scholar] [CrossRef]
- Xiao, S.; Nguyen, D.T.; Wu, B.; Hao, W. Genetic drift and indel mutation in the evolution of yeast mitochondrial genome size. Genome Biol. Evol. 2017, 9, 3088–3099. [Google Scholar] [CrossRef] [Green Version]
- Himmelstrand, K.; Olson, A.; Durling, M.B.M.; Karlsson, M.; Stenlid, J. Intronic and plasmid-derived regions contribute to the large mitochondrial genome sizes of agaricomycetes. Curr. Genet. 2014, 60, 303–313. [Google Scholar] [CrossRef] [Green Version]
- Gandini, C.L.; Sanchez-Puerta, M.V. Foreign plastid sequences in plant mitochondria are frequently acquired via mitochondrion-to-mitochondrion horizontal transfer. Sci. Rep. 2017, 7, 43402. [Google Scholar] [CrossRef]
- Galtier, N.; Roux, C.; Rousselle, M.; Romiguier, J.; Duret, L. Codon usage bias in animals: Disentangling the effects of natural selection, effective population size, and gc-biased gene conversion. Mol. Biol. Evol. 2018, 35, 1092–1103. [Google Scholar] [CrossRef] [Green Version]
- Bohlin, J.; Pettersson, J.H.O. Evolution of genomic base composition: From single cell microbes to multicellular animals. Computat. Struct. Biotec. 2019, 17, 362–370. [Google Scholar] [CrossRef]
- Yang, M.; Song, L.; Shi, Y.; Li, J.; Song, N. The first mitochondrial genome of the family epicopeiidae and higher-level phylogeny of Macroheterocera (Lepidoptera: Ditrysia). Int. J. Biol. Macromol. 2019, 136, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Sahyoun, A.H.; Bernt, M.; Stadler, P.F.; Tout, K. Gc skew and mitochondrial origins of replication. Mitochondrion 2014, 17, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Li, Y.; Kocot, K.M.; Yang, Y.; Qi, L.; Li, Q.; Halanych, K.M. Mitogenomics reveals phylogenetic relationships of Arcoida (Mollusca, Bivalvia) and multiple independent expansions and contractions in mitochondrial genome size. Mol. Phylogenet. Evol. 2020, 150, 106857. [Google Scholar] [CrossRef]
- Meng, X.; Zhao, N.; Shen, X.; Hao, J.; Liang, M.; Zhu, X.; Cheng, H.; Yan, B.; Liu, Z. Complete mitochondrial genome of Coelomactra Antiquata (Mollusca: Bivalvia): The first representative from the family mactridae with novel gene order and unusual tandem tepeats. Comp. Biochem. Physiol. Part D Genom. Proteom. 2012, 7, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Ojala, D.; Montoya, J.; Attardi, G. Trna punctuation model of rna processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Southworth, J.; Armitage, P.; Fallon, B.; Dawson, H.; Bryk, J.; Carr, M. Patterns of ancestral animal codon usage bias revealed through holozoan protists. Mol. Biol. Evol. 2018, 35, 2499–2511. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Meng, X.P.; Chu, K.H.; Zhao, N.N.; Tian, M.; Liang, M.; Hao, J. Comparative mitogenomic analysis reveals cryptic species: A case study in Mactridae (Mollusca: Bivalvia). Comp. Biochem. Physiol. Part D Genom. Proteom. 2014, 12, 1–9. [Google Scholar] [CrossRef]
- Cejp, B.; Ravara, A.; Aguado, M.T. First mitochondrial genomes of Chrysopetalidae (Annelida) from shallow-water and deep-sea chemosynthetic environments. Gene 2022, 815, 146159. [Google Scholar] [CrossRef]
- Wolstenholme, D. Genetic novelties in mitochondrial genomes of multicellular animals. Curr. Opin. Genet. Dev. 1992, 2, 918–925. [Google Scholar] [CrossRef]
- Wu, X.; Li, X.; Li, L.; Yu, Z. A unique trna gene family and a novel, highly expressed orf in the mitochondrial genome of the silver-lip pearl oyster, Pinctada Maxima (Bivalvia: Pteriidae). Gene 2012, 510, 22–31. [Google Scholar] [CrossRef]
- Sun, S.E.; Kong, L.; Yu, H.; Li, Q. Complete mitochondrial genome of Anadara vellicata (Bivalvia: Arcidae): A unique gene order and large atypical non-coding region. Comp. Biochem. Physiol. Part D Genom. Proteom. 2015, 16, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Jühling, F.; Pütz, J.; Bernt, M.; Donath, A.; Middendorf, M.; Florentz, C.; Stadler, P.F. Improved systematic trna gene annotation allows new insights into the evolution of mitochondrial trna structures and into the mechanisms of mitochondrial genome rearrangements. Nucleic Acids Res. 2012, 40, 2833–2845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, T.P.; Miya, M.; Mabuchi, K.; Nishida, M. Structure and variation of the mitochondrial genome of fishes. BMC Genom. 2016, 17, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowton, M.; Castro, L.R.; Austin, A.D. Mitochondrial gene rearrangements as phylogenetic characters in the invertebrates: The examination of genome ‘morphology’. Invertebr. Syst. 2002, 16, 345–356. [Google Scholar] [CrossRef]
- Alexandre, H.; Nelly, L.; Jean, D. Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of metazoa, and consequences for phylogenetic inferences. Syst. Biol. 2005, 54, 277–298. [Google Scholar] [CrossRef]
- Cameron, S.L.; Johnson, K.P.; Whiting, M.F. The mitochondrial genome of the screamer louse bothriometopus (Phthiraptera: Ischnocera): Effects of extensive gene rearrangements on the evolution of the genome. J. Mol. Evol. 2007, 65, 589–604. [Google Scholar] [CrossRef]
- Brown, M.W.M. Tandem duplications in animal mitochondrial dnas: Variation in incidence and gene content among lizards. Proc. Natl. Acad. Sci. USA 1987, 84, 7183–7187. [Google Scholar] [CrossRef] [Green Version]
- Lavrov, D.V.; Boore, J.L.; Brown, W.M. Complete mtdna sequences of two millipedes suggest a new model for mitochondrial gene rearrangements: Duplication and nonrandom loss. Mol. Biol. Evol. 2002, 19, 163–169. [Google Scholar] [CrossRef] [Green Version]
- Rokas, A.; Ladoukakis, E.; Zouros, E. Animal mitochondrial dna recombination revisited. Trends Ecol. Evol. 2003, 18, 411–417. [Google Scholar] [CrossRef]
- Cantatore, P.; Gadaleta, M.N.; Roberti, M.; Saccone, C.; Wilson, A.C. Duplication and remoulding of trna genes during the evolutionary rearrangement of mitochondrial genomes. Nature 1987, 329, 853–855. [Google Scholar] [CrossRef]
- Moritz, C.; Dowling, T.E.; Brown, W.M. Evolution of animal mitochondrial dna: Relevance for population biology and systematics. Annu. Rev. Ecol. Syst. 1987, 18, 269–292. [Google Scholar] [CrossRef]
- Dreyer, H.; Steiner, G. The complete sequence and gene organization of the mitochondrial genome of the gadilid scaphopod Siphonodentalium lobatum (Mollusca). Mol. Phylogenet. Evol. 2004, 31, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Lubośny, M.; Przyłucka, A.; Śmietanka, B.; Breton, S.; Burzyński, A. 2018 Actively transcribed and expressed atp8 gene in Mytilus edulis mussels. PeerJ6 2018, e4897. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Liu, H.; Zhang, H. Phylogeny and evolutionary radiation of the marine mussels (Bivalvia: Mytilidae) based on mitochondrial and nuclear genes. Mol. Phylogenet. Evol. 2018, 126, 233–240. [Google Scholar] [CrossRef]
- Samadi, S.; Quéméré, E.; Lorion, J.; Tillier, A.; von Cosel, R.; Lopez, P.; Cruaud, C.; Couloux, A.; Boisselier-Dubayle, M.C. Molecular phylogeny in mytilids supports the wooden steps to deep-sea vents hypothesis. Comptes Rendus Biol. 2007, 330, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhang, Y.; Xu, T.; Zhang, Y.; Mu, H.; Zhang, Y.; Lan, Y.; Fields, C.J.; Hui, J.H.L.; Zhang, W.; et al. Adaptation to deep-sea chemosynthetic environments as revealed by mussel genomes. Nat. Ecol. Evol. 2017, 1, 121. [Google Scholar] [CrossRef] [Green Version]
- Distel, D.L. Phylogenetic relationships among mytilidae (Bivalvia): 18S rrna data suggest convergence in mytilid body plans. Mol. Phylogenet. Evol. 2000, 15, 25–33. [Google Scholar] [CrossRef]
- Wood, A.R.; Apte, S.; MacAvoy, E.S.; Gardner, J.P. A molecular phylogeny of the marine mussel genus Perna (Bivalvia: Mytilidae) based on nuclear (its1&2) and mitochondrial (coi) dna sequences. Mol. Phylogenet. Evol. 2007, 44, 685–698. [Google Scholar] [CrossRef]
- Cunha, R.L.; Nicastro, K.R.; Costa, J.; McQuaid, C.D.; Serrão, E.A.; Zardi, G.I. Wider sampling reveals a non-sister relationship for geographically contiguous lineages of a marine mussel. Ecol. Evol. 2014, 4, 2070–2081. [Google Scholar] [CrossRef]
- Combosch, D.J.; Collins, T.M.; Glover, E.A.; Graf, D.L.; Bieler, R.A. Family-level tree of life for bivalves based on a sanger-sequencing approach. Mol. Phylogenet. Evol. 2017, 107, 191–208. [Google Scholar] [CrossRef] [Green Version]
- Trovant, B.; Orensanz, J.L.; Ruzzante, D.E.; Stotz, W.; Basso, N.G. Scorched mussels (Bivalvia: Mytilidae: Brachidontinae) from the temperate coasts of south America: Phylogenetic relationships, trans-pacific connections and the footprints of quaternary glaciations. Mol. Phylogenet. Evol. 2015, 82, 60–74. [Google Scholar] [CrossRef]
- Zhao, B.; Gao, S.; Zhao, M.; Lv, H.; Song, J.; Wang, H.; Zeng, Q.; Liu, J. Mitochondrial genomic analyses provide new insights into the "missing" atp8 and adaptive evolution of Mytilidae. BMC Genom. 2022, 23, 738. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M. Phylogenetic analysis of the subclass pteriomorphia (Bivalvia) from mtdna COI sequences. Mol. Phylogenet. Evol. 2003, 27, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Cunha, R.L.; Nicastro, K.R.; Zardi, G.I.; Madeira, C.; McQuaid, C.D.; Cox, C.J.; Castilho, R. Comparative mitogenomic analyses and gene rearrangements reject the alleged polyphyly of a bivalve genus. PeerJ 2022, 10, e13953. [Google Scholar] [CrossRef] [PubMed]
- Kafanov, A.I.; Drozdov, A.L. Comparative sperm morphology and phylogenetic classification of recent mytiloidea (Bivalvia). Malacologia 1998, 39, 129–139. [Google Scholar]
- Coan, E.V.; Valentich-Scott, P.; Bernard, F.R. Bivalve Seashells of Western North America: Marine Mollusks from Arctic Alaska to Baja California; Santa Barbara Museum of Natural History: Santa Barbara, CA, USA, 2000. [Google Scholar]
- Malchus, N. Constraints in the ligament ontogeny and evolution of pteriomorphian bivalvia. Palaeontology 2010, 47, 1539–1574. [Google Scholar] [CrossRef]
- Steiner, G.; Hammer, S. Molecular phylogeny of the bivalvia inferred from 18S rdna sequences with particular reference to the pteriomorphia. Geol. Soc. Lond. Spec. Publ. 2000, 177, 11–29. [Google Scholar] [CrossRef]
- Sun, W.; Gao, L. Phylogeny and comparative genomic analysis of pteriomorphia (Mollusca: Bivalvia) based on complete mitochondrial genomes. Mar. Biol. Res. 2017, 13, 255–268. [Google Scholar] [CrossRef]
- Danic-Tchaleu, G.; Heurtebise, S.; Morga, B.; Lapègue, S. Complete mitochondrial dna sequence of the European flat oyster Ostrea edulis confirms ostreidae classification. BMC Res. Notes 2011, 4, 400. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Size (bp) | A (%) | T (%) | G (%) | C (%) | A + T (%) | AT-Skew | GC-Skew |
|---|---|---|---|---|---|---|---|---|
| Mitogenome | 14,713 | 25.75 | 43.56 | 20.97 | 9.73 | 69.31 | –0.257 | 0.366 |
| cox1 | 1564 | 22.89 | 43.48 | 22.06 | 11.57 | 66.37 | –0.310 | 0.312 |
| cox2 | 687 | 24.89 | 41.48 | 22.56 | 11.06 | 60.70 | –0.250 | 0.342 |
| atp6 | 715 | 20.70 | 47.69 | 21.96 | 9.65 | 63.96 | –0.395 | 0.389 |
| atp8 | 122 | 20.93 | 52.71 | 23.26 | 3.10 | 73.64 | –0.431 | 0.765 |
| cox3 | 843 | 23.61 | 45.55 | 21.83 | 9.02 | 69.16 | –0.317 | 0.415 |
| nad3 | 348 | 23.28 | 47.70 | 20.69 | 8.33 | 70.98 | –0.344 | 0.426 |
| nad1 | 945 | 24.02 | 45.29 | 21.27 | 9.42 | 69.31 | –0.307 | 0.386 |
| nad5 | 1710 | 25.32 | 44.27 | 22.81 | 7.60 | 64.96 | –0.272 | 0.500 |
| nad4 | 1302 | 24.04 | 47.85 | 20.20 | 7.91 | 71.89 | –0.331 | 0.437 |
| nad4l | 273 | 26.74 | 42.86 | 22.71 | 7.69 | 69.60 | –0.232 | 0.494 |
| nad6 | 465 | 21.51 | 50.75 | 21.08 | 6.67 | 62.42 | –0.405 | 0.519 |
| cob | 1167 | 23.31 | 45.59 | 19.37 | 11.74 | 64.05 | –0.323 | 0.245 |
| nad2 | 1041 | 26.61 | 43.04 | 19.88 | 10.47 | 63.76 | –0.236 | 0.310 |
| tRNAs | 1435 | 30.66 | 37.84 | 20.07 | 11.43 | 68.50 | –0.105 | 0.274 |
| rRNAs | 1872 | 30.78 | 38.83 | 20.08 | 10.32 | 69.61 | –0.115 | 0.321 |
| PCGs | 11,189 | 23.94 | 45.27 | 21.36 | 9.43 | 69.21 | –0.597 | 0.387 |
| Gene | Strand | Location | Length | Codons | Intergenic Nucleotide (bp) | Anticodon | |
|---|---|---|---|---|---|---|---|
| Start | Stop | ||||||
| cox1 | + | 1 | 1564 | 1564 | ATA/T | 0 | |
| trnF | + | 1565 | 1631 | 67 | 0 | TTC | |
| rrnL | + | 1632 | 2709 | 1078 | 73 | ||
| trnY | + | 2783 | 2850 | 68 | 0 | TAC | |
| nad4l | + | 2851 | 3123 | 273 | ATG/TAA | 0 | |
| trnP | + | 3124 | 3189 | 66 | 0 | CCA | |
| trnE | + | 3190 | 3247 | 58 | 1 | GAA | |
| trnC | + | 3249 | 3312 | 64 | 0 | TGC | |
| nad3 | + | 3313 | 3660 | 348 | ATG/TAA | 5 | |
| trnW | + | 3666 | 3730 | 65 | 10 | TGA | |
| trnL2 | + | 3741 | 3805 | 65 | 2 | TTA | |
| trnK | + | 3808 | 3873 | 66 | 0 | AAA | |
| trnL1 | + | 3874 | 3939 | 66 | −4 | CTA | |
| trnG | + | 3936 | 3997 | 62 | 5 | GGA | |
| rrnS | + | 4003 | 4796 | 794 | 3 | ||
| trnQ | + | 4800 | 4867 | 68 | 0 | CAA | |
| trnH | + | 4868 | 4928 | 61 | 3 | CAC | |
| trnN | + | 4932 | 4996 | 65 | 0 | AAC | |
| trnM | + | 4997 | 5059 | 63 | 1 | ATG | |
| nad1 | + | 5061 | 6005 | 945 | ATG/TAG | 0 | |
| trnD | + | 6004 | 6071 | 68 | 1 | GAC | |
| nad2 | + | 6073 | 7113 | 1041 | ATG/TAA | 5 | |
| trnA | + | 7119 | 7184 | 66 | 0 | GCA | |
| trnV | + | 7185 | 7247 | 63 | 0 | GTA | |
| nad4 | + | 7248 | 8549 | 1302 | ATG/TAA | 42 | |
| cox3 | + | 8592 | 9434 | 843 | ATT/TAA | 0 | |
| trnS2 | + | 9435 | 9497 | 63 | 0 | TCA | |
| nad5 | + | 9498 | 11,207 | 1710 | ATG/TAG | 3 | |
| nad6 | + | 11,211 | 11,675 | 465 | ATG/TAA | 39 | |
| trnI | + | 11,715 | 11,779 | 65 | 3 | ATC | |
| trnT | + | 11,783 | 11,849 | 67 | 0 | ACA | |
| cob | + | 11,850 | 13,016 | 1167 | ATT/TAG | 5 | |
| cox2 | + | 13,022 | 13,708 | 687 | ATG/TAG | 5 | |
| trnR | + | 13,714 | 13,783 | 70 | 1 | CGA | |
| atp8 | + | 13,785 | 13,913 | 122 | GTG/TAA | −7 | |
| atp6 | + | 13,907 | 14,621 | 715 | ATG/T | 0 | |
| trnS1 | + | 14,622 | 14,690 | 69 | 23 | AGC | |
| Codon | Count | RSCU | Codon | Count | RSCU | Codon | Count | RSCU | Codon | Count | RSCU |
|---|---|---|---|---|---|---|---|---|---|---|---|
| UUU (F) | 399 | 1.58 | UCU (S) | 73 | 1.43 | UAU (Y) | 213 | 1.67 | UGU (C) | 145 | 1.6 |
| UUC (F) | 106 | 0.42 | UCC (S) | 26 | 0.51 | UAC (Y) | 42 | 0.33 | UGC (C) | 36 | 0.40 |
| UUA (L) | 282 | 2.16 | UCA (S) | 46 | 0.90 | UAA (*) | 172 | 1.16 | UGA (W) | 112 | 1.09 |
| UUG (L) | 206 | 1.58 | UCG (S) | 25 | 0.49 | UAG (*) | 124 | 0.84 | UGG (W) | 93 | 0.91 |
| CUU (L) | 124 | 0.95 | CCU (P) | 38 | 2.49 | CAU (H) | 56 | 1.78 | CGU (R) | 34 | 1.97 |
| CUC (L) | 29 | 0.22 | CCC (P) | 6 | 0.39 | CAC (H) | 7 | 0.22 | CGC (R) | 11 | 0.64 |
| CUA (L) | 74 | 0.57 | CCA (P) | 8 | 0.52 | CAA (Q) | 27 | 1.00 | CGA (R) | 13 | 0.75 |
| CUG (L) | 67 | 0.51 | CCG (P) | 9 | 0.59 | CAG (Q) | 27 | 1.00 | CGG (R) | 11 | 0.64 |
| AUU (I) | 226 | 1.65 | ACU (T) | 49 | 2.28 | AAU (N) | 98 | 1.41 | AGU (S) | 88 | 1.73 |
| AUC (I) | 48 | 0.35 | ACC (T) | 8 | 0.37 | AAC (N) | 41 | 0.59 | AGC (S) | 21 | 0.41 |
| AUA (M) | 147 | 1.08 | ACA (T) | 15 | 0.70 | AAA (K) | 114 | 1.19 | AGA (S) | 69 | 1.36 |
| AUG (M) | 125 | 0.92 | ACG (T) | 14 | 0.65 | AAG (K) | 77 | 0.81 | AGG (S) | 59 | 1.16 |
| GUU (V) | 203 | 1.82 | GCU (A) | 55 | 2.06 | GAU (D) | 98 | 1.68 | GGU (G) | 130 | 1.99 |
| GUC (V) | 41 | 0.37 | GCC (A) | 12 | 0.45 | GAC (D) | 19 | 0.32 | GGC (G) | 13 | 0.20 |
| GUA (V) | 100 | 0.89 | GCA (A) | 22 | 0.82 | GAA (E) | 83 | 1.26 | GGA (G) | 58 | 0.89 |
| GUG (V) | 103 | 0.92 | GCG (A) | 18 | 0.67 | GAG (E) | 49 | 0.74 | GGG (G) | 60 | 0.92 |
| Species | Position (bp) | Size (bp) | Intergenic Region (bp) | Start Codon | Stop Codon | GenBank |
|---|---|---|---|---|---|---|
| Modiolus kurilensis | 676–861 | 186 | 191 | ATG | TAA | KY242717 |
| Modiolus modiolus | 3240–3419 | 180 | 194 | ATG | TAA | NC_033537 |
| Modiolus nipponicus | 3409–3636 | 228 | 304 | ATG | TAA | NC_044130 |
| Modiolus philippinarum | 16,113–16,304 | 192 | 209 | ATG | TAA | NC_035422 |
| Modiolus comptus | 3106–3276 | 171 | 670 | ATG | TAG | MN602036 |
| Arcuatula senhousia | 7403–7594 | 192 | 216 | ATG | TAG | NC_014590 |
| Mytilus unguiculatus | 8677–8949 | 273 | 286 | ATG | TAA | NC_024733 |
| Mytilisepta keenae | 15,718–15,882 | 165 | 186 | ATG | TAA | MK721542 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, M.; Gu, Z.; Huang, J.; Guo, B.; Jiang, L.; Xu, K.; Ye, Y.; Li, J. The Complete Mitochondrial Genome of Mytilisepta virgata (Mollusca: Bivalvia), Novel Gene Rearrangements, and the Phylogenetic Relationships of Mytilidae. Genes 2023, 14, 910. https://doi.org/10.3390/genes14040910
Xu M, Gu Z, Huang J, Guo B, Jiang L, Xu K, Ye Y, Li J. The Complete Mitochondrial Genome of Mytilisepta virgata (Mollusca: Bivalvia), Novel Gene Rearrangements, and the Phylogenetic Relationships of Mytilidae. Genes. 2023; 14(4):910. https://doi.org/10.3390/genes14040910
Chicago/Turabian StyleXu, Minhui, Zhongqi Gu, Ji Huang, Baoying Guo, Lihua Jiang, Kaida Xu, Yingying Ye, and Jiji Li. 2023. "The Complete Mitochondrial Genome of Mytilisepta virgata (Mollusca: Bivalvia), Novel Gene Rearrangements, and the Phylogenetic Relationships of Mytilidae" Genes 14, no. 4: 910. https://doi.org/10.3390/genes14040910