
An Improved Machine Learning-Based Approach to Assess the Microbial Diversity in Major North Indian River Ecosystems

 ,
,  , ,
, ,  and
and 
Abstract
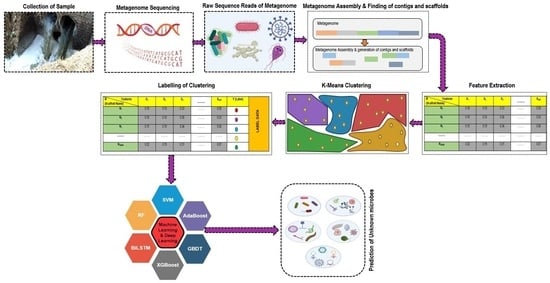
:
1. Introduction
2. Materials and Methods
2.1. Data Acquisition
2.2. Preprocessing and Genome Assembly
2.3. Feature Extraction
2.3.1. CTD (Composition, Transition and Distribution) Feature
2.3.2. Fragment Composition Feature
2.3.3. Gap-Pair Composition Feature
2.3.4. CKSAAP (Composition of K-Spaced Amino Acid Pairs) Features
2.4. Clustering for Binning
2.4.1. K-Means Clustering
2.4.2. Evaluation of Clusters
2.5. Machine Learning for Classification
2.5.1. Support Vector Machine
2.5.2. Random Forest
2.5.3. Gradient Boosting Decision Tree
2.5.4. XGBoost
2.5.5. AdaBoost
2.5.6. BiLSTM
2.6. Training and Validation
2.7. Comparison with the State-of-the-art Tools
3. Results
3.1. The Metagenome Assembly
3.2. Diversity of Microbial Population
3.3. Binning of Metagenomic Data
3.4. Performance of Machine Learning Algorithms at Different Locations
3.4.1. Overall Prediction Performance
3.4.2. Class-Wise Prediction Performance
3.5. Comparison of Different Classification Models
3.6. Comparison with the State-of-the-Art Tools
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cullen, C.M.; Aneja, K.K.; Beyhan, S.; Cho, C.E.; Woloszynek, S.; Convertino, M.; McCoy, S.J.; Zhang, Y.; Anderson, M.Z.; Alvarez-Ponce, D.; et al. Emerging Priorities for Microbiome Research. Front. Microbiol. 2020, 11, 136. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.P.; Ghazi, M.G.; Katdare, S.; Dasgupta, N.; Mondol, S.; Gupta, S.K.; Hussain, S.A. Microsatellite Analysis Reveals Low Genetic Diversity in Managed Populations of the Critically Endangered Gharial (Gavialis Gangeticus) in India. Sci. Rep. 2021, 11, 5627. [Google Scholar] [CrossRef] [PubMed]
- Ghurye, J.S.; Cepeda-Espinoza, V.; Pop, M. Focus: Microbiome: Metagenomic Assembly: Overview, Challenges and Applications. Yale J. Biol. Med. 2016, 89, 353. [Google Scholar] [PubMed]
- Handelsman, J. Metagenomics: Application of Genomics to Uncultured Microorganisms. Microbiol. Mol. Biol. Rev. 2005, 69, 195. [Google Scholar] [CrossRef]
- Strous, M.; Kraft, B.; Bisdorf, R.; Tegetmeyer, H.E. The Binning of Metagenomic Contigs for Microbial Physiology of Mixed Cultures. Front. Microbiol. 2012, 3, 410. [Google Scholar] [CrossRef]
- Wu, Y.W.; Ye, Y. A novel abundance-based algorithm for binning metagenomic sequences using l-tuples. J. Comput. Biol. 2011, 18, 523–534. [Google Scholar] [CrossRef]
- Mavromatis, K.; Ivanova, N.; Barry, K.; Shapiro, H.; Goltsman, E.; McHardy, A.C.; Kyrpides, N.C. Use of simulated data sets to evaluate the fidelity of metagenomic processing methods. Nat. Methods 2007, 4, 495–500. [Google Scholar] [CrossRef]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN Analysis of Metagenomic Data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef]
- McHardy, A.C.; Martín, H.G.; Tsirigos, A.; Hugenholtz, P.; Rigoutsos, I. Accurate Phylogenetic Classification of Variable-Length DNA Fragments. Nat. Methods 2007, 4, 63–72. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Reddy, R.M.; Mohammed, M.H.; Mande, S.S. MetaCAA: A Clustering-Aided Methodology for Efficient Assembly of Metagenomic Datasets. Genomics 2014, 103, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Kanaya, S.; Kinouchi, M.; Ichiba, Y.; Kozuki, T.; Ikemura, T. Informatics for Unveiling Hidden Genome Signatures. Genome Res. 2003, 13, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Dhungel, E.; Mreyoud, Y.; Gwak, H.J.; Rajeh, A.; Rho, M.; Ahn, T.H. MegaR: An Interactive R Package for Rapid Sample Classification and Phenotype Prediction Using Metagenome Profiles and Machine Learning. BMC Bioinform. 2021, 22, 25. [Google Scholar] [CrossRef] [PubMed]
- Pasolli, E.; Truong, D.T.; Malik, F.; Waldron, L.; Segata, N. Machine Learning Meta-Analysis of Large Metagenomic Datasets: Tools and Biological Insights. PLoS Comput. Biol. 2016, 12, e1004977. [Google Scholar] [CrossRef] [PubMed]
- Behera, B.K.; Patra, B.; Chakraborty, H.J.; Sahu, P.; Rout, A.K.; Sarkar, D.J.; Parida, P.K.; Raman, R.K.; Rao, A.R.; Rai, A.; et al. Metagenome Analysis from the Sediment of River Ganga and Yamuna: In Search of Beneficial Microbiome. PLoS ONE 2020, 15, e0239594. [Google Scholar] [CrossRef]
- Sahu, N.; Deori, M.; Ghosh, I. Metagenomics Study of Contaminated Sediments from the Yamuna River at Kalindi Kunj, Delhi, India. Genome Announc. 2018, 6, e01379-17. [Google Scholar] [CrossRef]
- Behera, B.K.; Chakraborty, H.J.; Patra, B.; Rout, A.K.; Dehury, B.; Das, B.K.; Sarkar, D.J.; Parida, P.K.; Raman, R.K.; Rao, A.R.; et al. Metagenomic Analysis Reveals Bacterial and Fungal Diversity and Their Bioremediation Potential from Sediments of River Ganga and Yamuna in India. Front. Microbiol. 2020, 11, 2531. [Google Scholar] [CrossRef]
- Samson, R.; Shah, M.; Yadav, R.; Sarode, P.; Rajput, V.; Dastager, S.G.; Dharne, M.S.; Khairnar, K. Metagenomic Insights to Understand Transient Influence of Yamuna River on Taxonomic and Functional Aspects of Bacterial and Archaeal Communities of River Ganges. Sci. Total Environ. 2019, 674, 288–299. [Google Scholar] [CrossRef]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and Sensitive Taxonomic Classification for Metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef]
- Ghannam, R.B.; Techtmann, S.M. Machine Learning Applications in Microbial Ecology, Human Microbiome Studies, and Environmental Monitoring. Comput. Struct. Biotechnol. J. 2021, 19, 1092–1107. [Google Scholar] [CrossRef] [PubMed]
- McCoy, R.C.; Taylor, R.W.; Blauwkamp, T.A.; Kelley, J.L.; Kertesz, M.; Pushkarev, D.; Petrov, D.A.; Fiston-Lavier, A.S. Illumina TruSeq Synthetic Long-Reads Empower de Novo Assembly and Resolve Complex, Highly-Repetitive Transposable Elements. PLoS ONE 2014, 9, e106689. [Google Scholar] [CrossRef]
- FastQC: A Quality Control Tool for High Throughput Sequence Data—ScienceOpen. Available online: https://www.scienceopen.com/document?vid=de674375-ab83-4595-afa9-4c8aa9e4e736 (accessed on 6 August 2022).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An Empirically Improved Memory-Efficient Short-Read de Novo Assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Pruitt, K.D.; Schoch, C.L.; Sherry, S.T.; Karsch-Mizrachi, I. GenBank. Nucleic Acids Res. 2021, 49, D92–D96. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Using the BioSeqClass Package; Shanghai Institutes for Biological Sciences: Shanghai, China, 2017. [Google Scholar]
- Keck, F. Handling Biological Sequences in R with the Bioseq Package. Methods Ecol. Evol. 2020, 11, 1728–1732. [Google Scholar] [CrossRef]
- Charif, D.; Lobry, J.R. SeqinR 1.0-2: A Contributed Package to the R Project for Statistical Computing Devoted to Biological Sequences Retrieval and Analysis. In Structural Approaches to Sequence Evolution: Molecules, Networks, Populations, 207–232; Springer: Berlin/Heidelberg, Germany, 2007. [Google Scholar] [CrossRef]
- Biostrings: Efficient Manipulation of Biological Strings Version 2.58.0 from Bioconductor. Available online: https://rdrr.io/bioc/Biostrings/ (accessed on 6 August 2022).
- Cai, C.Z.; Han, L.Y.; Ji, Z.L.; Chen, X.; Chen, Y.Z. SVM-Prot: Web-Based Support Vector Machine Software for Functional Classification of a Protein from Its Primary Sequence. Nucleic Acids Res. 2003, 31, 3692–3697. [Google Scholar] [CrossRef]
- Yu, C.-S.; Lin, C.-J.; Hwang, J.-K. Predicting Subcellular Localization of Proteins for Gram-Negative Bacteria by Support Vector Machines Based on n-Peptide Compositions. Protein Sci. 2004, 13, 1402–1406. [Google Scholar] [CrossRef]
- Yu, C.S.; Chen, Y.C.; Lu, C.H.; Hwang, J.K. Prediction of Protein Subcellular Localization. Proteins 2006, 64, 643–651. [Google Scholar] [CrossRef]
- Chen, Y.Z.; Tang, Y.R.; Sheng, Z.Y.; Zhang, Z. Prediction of Mucin-Type O-Glycosylation Sites in Mammalian Proteins Using the Composition of k-Spaced Amino Acid Pairs. BMC Bioinform. 2008, 9, 101. [Google Scholar] [CrossRef] [PubMed]
- Team, R.C. A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria; Scientific Research Publishing: Wuhan, China, 2013; Available online: https://www.scirp.org/(S(i43dyn45teexjx455qlt3d2q))/reference/ReferencesPapers.aspx?ReferenceID=1778705 (accessed on 6 August 2022).
- Dokmanic, I.; Parhizkar, R.; Ranieri, J.; Vetterli, M. Euclidean distance matrices: Essential theory, algorithms, and applications. IEEE Signal Process. Mag. 2015, 32, 12–30. [Google Scholar] [CrossRef]
- Shahapure, K.R.; Nicholas, C. Cluster Quality Analysis Using Silhouette score. In Proceedings of the 2020 IEEE 7th International Conference on Data Science and Advanced Analytics (DSAA), Sydney, Australia, 6–9 October 2020; p. 747. [Google Scholar]
- Boser, B.E.; Guyon, I.M.; Vapnik, V.N. A Training Algorithm for Optimal Margin Classifiers. In Proceedings of the Fifth Annual Workshop on Computational Learning Theory-COLT ‘92, Pittsburgh, PA, USA, 27–29 July 1992. [Google Scholar] [CrossRef]
- Vapnik, V.N. The Nature of Statistical Learning Theory; Springer Science and Business Media LLC: New York, NY, USA, 2000. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Breiman, L. Bagging Predictors. Mach. Learn. 1996, 24, 123–140. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Ho, T.K. Random Decision Forest. In Proceedings of the 3rd International Conference on Document Analysis and Recognition, Montreal, QC, Canada, 14–16 August 1995; References-Scientific Research Publishing: Wuhan, China, 1995; pp. 278–282. Available online: https://www.scirp.org/(S(i43dyn45teexjx455qlt3d2q))/reference/ReferencesPapers.aspx?ReferenceID=1698778 (accessed on 6 August 2022).
- Friedman, J.H. Greedy Function Approximation: A Gradient Boosting Machine. Ann. Statist. 2001, 29, 1189–1232. [Google Scholar] [CrossRef]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar] [CrossRef]
- Freund, Y.; Schapire, R.E. A Decision-Theoretic Generalization of On-Line Learning and an Application to Boosting. J. Comput. Syst. Sci. 1997, 55, 119–139. [Google Scholar] [CrossRef]
- Aziz Sharfuddin, A.; Nafis Tihami, M.; Saiful Islam, M. A Deep Recurrent Neural Network with BiLSTM Model for Sentiment Classification. In Proceedings of the 2018 International Conference on Bangla Speech and Language Processing, Sylhet, Bangladesh, 21–22 September 2018. [Google Scholar] [CrossRef]
- Feurer, M.; Klein, A.; Eggensperger, K.; Springenberg, J.T.; Blum, M.; Hutter, F. Auto-Sklearn: Efficient and Robust Automated Machine Learning. In Automated Machine Learning; The Springer Series on Challenges in Machine Learning; Springer: Cham, Switzerland, 2019; pp. 113–134. [Google Scholar] [CrossRef]
- Brownlee, J. A Gentle Introduction to K-Fold Cross-Validation. Available online: https://machinelearningmastery.com/k-fold-cross-validation/ (accessed on 6 August 2022).
- Kohavi, R.; Provost, F. Glossary of Terms. Machine Learning—Special Issue on Applications of Machine Learning and the Knowledge Discovery Process. Mach. Learn. 1998, 30, 271–274. Available online: https://www.scirp.org/(S(czeh2tfqyw2orz553k1w0r45))/reference/ReferencesPapers.aspx?ReferenceID=2264480 (accessed on 11 August 2022).
- Chandran, H.; Meena, M.; Sharma, K. Microbial Biodiversity and Bioremediation Assessment Through Omics Approaches. Front. Environ. Chem. 2020, 1, 570326. [Google Scholar] [CrossRef]
- Trinh, P.; Zaneveld, J.R.; Safranek, S.; Rabinowitz, P.M. One Health Relationships Between Human, Animal, and Environmental Microbiomes: A Mini-Review. Front. Public Health 2018, 6, 235. [Google Scholar] [CrossRef]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- Simpson, E.H. Measurement of diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Köchling, T.; Sanz, J.L.; Galdino, L.; Florencio, L.; Kato, M.T. Impact of Pollution on the Microbial Diversity of a Tropical River in an Urbanized Region of Northeastern Brazil. Int. Microbiol. 2017, 20, 11–24. [Google Scholar] [CrossRef]
- Katiyar, S. Impact of Tannery Effluent with Special Reference to Seasonal Variation on Physico-Chemical Characteristics of River Water at Kanpur (U.P), India. J. Environ. Anal. Toxicol. 2011, 1, 4. [Google Scholar] [CrossRef]
- Chowdhury, M.; Mostafa, M.G.; Biswas, T.K.; Mandal, A.; Saha, A.K. Characterization of the Effluents from Leather Processing Industries. Environ. Process. 2015, 2, 173–187. [Google Scholar] [CrossRef]
- Fitzgerald, M.; Moses, L.; Kawano, A. At the Cross Roads. Ed. Publ. 2003, 136, 1–35. [Google Scholar] [CrossRef]
- Wang, J.; Fan, H.; He, X.; Zhang, F.; Xiao, J.; Yan, Z.; Feng, J.; Li, R. Response of Bacterial Communities to Variation in Water Quality and Physicochemical Conditions in a River-Reservoir System. Glob. Ecol. Conserv. 2021, 27, e01541. [Google Scholar] [CrossRef]
- Oulas, A.; Pavloudi, C.; Polymenakou, P.; Pavlopoulos, G.A.; Papanikolaou, N.; Kotoulas, G.; Arvanitidis, C.; Iliopoulos, I. Metagenomics: Tools and Insights for Analyzing next-Generation Sequencing Data Derived from Biodiversity Studies. Bioinform. Biol. Insights 2015, 9, 75–88. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classifiers | Parameters | ||
|---|---|---|---|
| SVM | C = 10, gamma = 0.001, kernel = ‘rbf’ | ||
| RF | max_depth = 25, min_samples_leaf = 2, n_estimators = 25, random_state = 42 | ||
| GBDT | n_splits = 10, n_repeats = 3, random_state = 1 | ||
| XGBoost | learning_rate = 0.2, n_estimators = 1000, max_depth = 4,min_child_weight = 6, gamma = 0, subsample = 0.8, colsample_bytree = 0.8, objective = ‘binary:logistic’,nthread = 4, scale_pos_weight = 1, seed = 27 | ||
| AdaBoost | learning_rate = 0.1, n_estimators=100 | ||
| BiLSTM | Layer (type) | Output Shape | Param |
| bidirectional (Bidirectional) | (None, 30, 128) | 164,864 | |
| bidirectional_1 (Bidirectional) | (None, 128) | 98,816 | |
| dense (Dense) | (None, 1) | 129 | |
| Total params: 263,809 Trainable params: 263,809 Non-trainable params: 0 | |||
| Parameters | Farakka | Kanpur | Delhi |
|---|---|---|---|
| No. of raw reads | 111,809,841 | 98,019,576 | 195,606,736 |
| No. of trimmed reads | 108,526,822 | 96,309,665 | 192,296,837 |
| Number of scaffolds | 9771 | 83,982 | 69,083 |
| Number of contigs | 44,132,563 | 18,200,983 | 54,832,125 |
| Total scaffold length(bp) | 4,232,650 | 49,399,583 | 36,990,293 |
| Average scaffold length | 433 | 588 | 535 |
| Longest scaffold (bp) | 3859 | 7427 | 5265 |
| N50 | 128 | 110 | 128 |
| N90 | 95 | 91 | 96 |
| Microbial Diversity | Ganga | Yamuna | ||
|---|---|---|---|---|
| Farakka | Kanpur | Delhi | ||
| Cellular | Bacteria | Streptomyces(400), Pseudomonas(180), Sphingomonas(180), Bradyrhizobium(140), Burkholderia(130), Azospirillum(100) | Pseudomonas(2980), Streptomyces(2910), Burkholderia(1370), Sphingomonas(1320), Bradyrhizobium(1280) | Dechloromonas(3095), Paraburkholderia(2047), Streptomyces(2012), Achromobacter(1359), Acidobacteria(1125), Aeoliella(1063) |
| Archaea | Natrinema (258), Nitrososphaera(197), Cenarchaeum(125), Thermococcus(95), Salinigranum(54) | Haloterrigena (395), Aeropyrum (187), Haloarcula(135), Methanothrix(123) | Thermococcus(348), Haloferax(295), Halobacterium(181), Halogeometricum(126) | |
| Eukaryota | Paracoccus(147), Lobosporangium(159), Parastrongyloides(57), Emiliania(38) | Cyclotella(356), Timema(329), Aspergillus(259) | Gaeumannomyces(124), Phoenix(98), Strongyloides(56) | |
| Non-cellular | Virus | Variovorax(45), Roseovarius(21) | Variovorax(347), Roseovarius(321) | Roseovarius(124), Variovorax(119) |
| Classifiers | Dataset (Location) | Accuracy | Precision | Recall | F1 Score | AUC–ROC |
|---|---|---|---|---|---|---|
| SVM | Farakka | 0.76 | 0.80 | 0.75 | 0.80 | 0.81 |
| Kanpur | 0.78 | 0.78 | 0.78 | 0.78 | 0.83 | |
| Delhi | 0.86 | 0.81 | 0.79 | 0.79 | 0.86 | |
| RF | Farakka | 0.85 | 0.83 | 0.76 | 0.80 | 0.85 |
| Kanpur | 0.84 | 0.79 | 0.79 | 0.79 | 0.80 | |
| Delhi | 0.88 | 0.85 | 0.80 | 0.80 | 0.87 | |
| GBDT | Farakka | 0.80 | 0.80 | 0.78 | 0.80 | 0.79 |
| Kanpur | 0.80 | 0.80 | 0.82 | 0.79 | 0.80 | |
| Delhi | 0.85 | 0.81 | 0.79 | 0.80 | 0.81 | |
| XGBoost | Farakka | 0.85 | 0.79 | 0.77 | 0.79 | 0.75 |
| Kanpur | 0.79 | 0.76 | 0.73 | 0.79 | 0.81 | |
| Delhi | 0.80 | 0.83 | 0.79 | 0.80 | 0.79 | |
| AdaBoost | Farakka | 0.77 | 0.78 | 0.77 | 0.80 | 0.78 |
| Kanpur | 0.79 | 0.78 | 0.79 | 0.77 | 0.74 | |
| Delhi | 0.80 | 0.84 | 0.81 | 0.80 | 0.77 | |
| BiLSTM | Farakka | 0.81 | 0.80 | 0.75 | 0.79 | 0.75 |
| Kanpur | 0.75 | 0.79 | 0.80 | 0.80 | 0.79 | |
| Delhi | 0.79 | 0.80 | 0.78 | 0.78 | 0.76 |
| Classifiers | Multiclass | Accuracy | Precision | Recall | F1 Score | AUC–ROC |
|---|---|---|---|---|---|---|
| SVM | Bacteria | 0.75 | 0.80 | 0.75 | 0.79 | 0.74 |
| Archaea | 0.58 | 0.78 | 0.76 | 0.78 | 0.72 | |
| Eukaryota | 0.65 | 0.75 | 0.71 | 0.69 | 0.81 | |
| Virus | 0.76 | 0.80 | 0.75 | 0.80 | 0.80 | |
| Others | 0.66 | 0.76 | 0.74 | 0.80 | 0.78 | |
| RF | Bacteria | 0.70 | 0.76 | 0.68 | 0.80 | 0.85 |
| Archaea | 0.72 | 0.72 | 0.76 | 0.79 | 0.85 | |
| Eukaryota | 0.69 | 0.78 | 0.62 | 0.78 | 0.72 | |
| Virus | 0.85 | 0.83 | 0.77 | 0.80 | 0.80 | |
| Others | 0.68 | 0.71 | 0.77 | 0.79 | 0.75 | |
| GBDT | Bacteria | 0.69 | 0.80 | 0.78 | 0.80 | 0.59 |
| Archaea | 0.58 | 0.68 | 0.82 | 0.79 | 0.68 | |
| Eukaryota | 0.74 | 0.78 | 0.76 | 0.78 | 0.72 | |
| Virus | 0.80 | 0.80 | 0.78 | 0.80 | 0.79 | |
| Others | 0.77 | 0.76 | 0.74 | 0.80 | 0.78 | |
| XGBoost | Bacteria | 0.72 | 0.79 | 0.77 | 0.79 | 0.75 |
| Archaea | 0.79 | 0.76 | 0.73 | 0.69 | 0.61 | |
| Eukaryota | 0.69 | 0.68 | 0.78 | 0.80 | 0.75 | |
| Virus | 0.82 | 0.79 | 0.77 | 0.79 | 0.75 | |
| Others | 0.74 | 0.78 | 0.76 | 0.78 | 0.52 | |
| AdaBoost | Bacteria | 0.77 | 0.78 | 0.77 | 0.80 | 0.78 |
| Archaea | 0.61 | 0.78 | 0.79 | 0.77 | 0.64 | |
| Eukaryota | 0.69 | 0.75 | 0.65 | 0.77 | 0.78 | |
| Virus | 0.71 | 0.84 | 0.81 | 0.80 | 0.67 | |
| Others | 0.74 | 0.78 | 0.76 | 0.78 | 0.52 | |
| BiLSTM | Bacteria | 0.52 | 0.80 | 0.75 | 0.79 | 0.75 |
| Archaea | 0.75 | 0.79 | 0.75 | 0.71 | 0.72 | |
| Eukaryota | 0.82 | 0.78 | 0.77 | 0.79 | 0.70 | |
| Virus | 0.79 | 0.80 | 0.78 | 0.78 | 0.71 | |
| Others | 0.64 | 0.79 | 0.65 | 0.79 | 0.74 |
| Classifiers | Multiclass | Accuracy | Precision | Recall | F1 Score | AUC–ROC |
|---|---|---|---|---|---|---|
| SVM | Bacteria | 0.79 | 0.81 | 0.75 | 0.80 | 0.74 |
| Archaea | 0.74 | 0.78 | 0.76 | 0.78 | 0.72 | |
| Eukaryota | 0.78 | 0.75 | 0.71 | 0.69 | 0.80 | |
| Virus | 0.78 | 0.78 | 0.78 | 0.79 | 0.86 | |
| Others | 0.77 | 0.76 | 0.74 | 0.80 | 0.78 | |
| RF | Bacteria | 0.78 | 0.76 | 0.76 | 0.78 | 0.85 |
| Archaea | 0.71 | 0.79 | 0.71 | 0.79 | 0.80 | |
| Eukaryota | 0.74 | 0.78 | 0.76 | 0.78 | 0.72 | |
| Virus | 0.80 | 0.70 | 0.69 | 0.73 | 0.87 | |
| Others | 0.70 | 0.79 | 0.73 | 0.71 | 0.75 | |
| GBDT | Bacteria | 0.69 | 0.80 | 0.78 | 0.78 | 0.79 |
| Archaea | 0.75 | 0.80 | 0.82 | 0.79 | 0.80 | |
| Eukaryota | 0.74 | 0.78 | 0.76 | 0.78 | 0.72 | |
| Virus | 0.81 | 0.80 | 0.79 | 0.70 | 0.78 | |
| Others | 0.77 | 0.76 | 0.74 | 0.72 | 0.78 | |
| XGBoost | Bacteria | 0.72 | 0.75 | 0.69 | 0.79 | 0.75 |
| Archaea | 0.79 | 0.76 | 0.73 | 0.79 | 0.81 | |
| Eukaryota | 0.69 | 0.68 | 0.73 | 0.79 | 0.79 | |
| Virus | 0.78 | 0.63 | 0.71 | 0.70 | 0.79 | |
| Others | 0.74 | 0.71 | 0.59 | 0.72 | 0.72 | |
| AdaBoost | Bacteria | 0.77 | 0.78 | 0.77 | 0.71 | 0.71 |
| Archaea | 0.81 | 0.78 | 0.79 | 0.77 | 0.74 | |
| Eukaryota | 0.69 | 0.75 | 0.65 | 0.77 | 0.70 | |
| Virus | 0.80 | 0.78 | 0.78 | 0.75 | 0.69 | |
| Others | 0.74 | 0.78 | 0.76 | 0.70 | 0.72 | |
| BiLSTM | Bacteria | 0.62 | 0.68 | 0.75 | 0.79 | 0.75 |
| Archaea | 0.75 | 0.79 | 0.80 | 0.80 | 0.79 | |
| Eukaryota | 0.71 | 0.78 | 0.77 | 0.80 | 0.78 | |
| Virus | 0.73 | 0.77 | 0.78 | 0.78 | 0.76 | |
| Others | 0.64 | 0.72 | 0.65 | 0.79 | 0.74 |
| Classifiers | Multiclass | Accuracy | Precision | Recall | F1 Score | AUC–ROC |
|---|---|---|---|---|---|---|
| SVM | Bacteria | 0.71 | 0.80 | 0.75 | 0.79 | 0.74 |
| Archaea | 0.74 | 0.78 | 0.76 | 0.78 | 0.72 | |
| Eukaryota | 0.80 | 0.75 | 0.71 | 0.69 | 0.80 | |
| Virus | 0.82 | 0.81 | 0.79 | 0.79 | 0.86 | |
| Others | 0.77 | 0.76 | 0.74 | 0.80 | 0.78 | |
| RF | Bacteria | 0.78 | 0.76 | 0.76 | 0.80 | 0.85 |
| Archaea | 0.80 | 0.79 | 0.79 | 0.79 | 0.80 | |
| Eukaryota | 0.74 | 0.78 | 0.76 | 0.78 | 0.72 | |
| Virus | 0.84 | 0.85 | 0.78 | 0.80 | 0.87 | |
| Others | 0.82 | 0.79 | 0.77 | 0.79 | 0.75 | |
| GBDT | Bacteria | 0.69 | 0.80 | 0.78 | 0.70 | 0.79 |
| Archaea | 0.55 | 0.80 | 0.82 | 0.79 | 0.80 | |
| Eukaryota | 0.74 | 0.78 | 0.76 | 0.78 | 0.72 | |
| Virus | 0.81 | 0.81 | 0.79 | 0.80 | 0.81 | |
| Others | 0.77 | 0.76 | 0.74 | 0.80 | 0.78 | |
| XGBoost | Bacteria | 0.72 | 0.79 | 0.77 | 0.79 | 0.75 |
| Archaea | 0.79 | 0.76 | 0.73 | 0.79 | 0.81 | |
| Eukaryota | 0.69 | 0.80 | 0.78 | 0.80 | 0.79 | |
| Virus | 0.81 | 0.83 | 0.79 | 0.80 | 0.79 | |
| Others | 0.74 | 0.78 | 0.76 | 0.78 | 0.72 | |
| AdaBoost | Bacteria | 0.77 | 0.78 | 0.77 | 0.80 | 0.78 |
| Archaea | 0.80 | 0.78 | 0.79 | 0.77 | 0.74 | |
| Eukaryota | 0.69 | 0.75 | 0.65 | 0.77 | 0.79 | |
| Virus | 0.80 | 0.84 | 0.81 | 0.80 | 0.77 | |
| Others | 0.74 | 0.78 | 0.76 | 0.78 | 0.72 | |
| BiLSTM | Bacteria | 0.78 | 0.80 | 0.75 | 0.72 | 0.75 |
| Archaea | 0.75 | 0.79 | 0.78 | 0.78 | 0.72 | |
| Eukaryota | 0.77 | 0.78 | 0.77 | 0.78 | 0.76 | |
| Virus | 0.79 | 0.80 | 0.78 | 0.78 | 0.76 | |
| Others | 0.64 | 0.79 | 0.65 | 0.75 | 0.70 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choudhury, N.; Sahu, T.K.; Rao, A.R.; Rout, A.K.; Behera, B.K. An Improved Machine Learning-Based Approach to Assess the Microbial Diversity in Major North Indian River Ecosystems. Genes 2023, 14, 1082. https://doi.org/10.3390/genes14051082
Choudhury N, Sahu TK, Rao AR, Rout AK, Behera BK. An Improved Machine Learning-Based Approach to Assess the Microbial Diversity in Major North Indian River Ecosystems. Genes. 2023; 14(5):1082. https://doi.org/10.3390/genes14051082
Chicago/Turabian StyleChoudhury, Nalinikanta, Tanmaya Kumar Sahu, Atmakuri Ramakrishna Rao, Ajaya Kumar Rout, and Bijay Kumar Behera. 2023. "An Improved Machine Learning-Based Approach to Assess the Microbial Diversity in Major North Indian River Ecosystems" Genes 14, no. 5: 1082. https://doi.org/10.3390/genes14051082