

Comparisons of Chloroplast Genome Mutations among 13 Samples of Oil-Tea Camellia from South China

Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Materials
2.2. SNP and InDel Analysis
2.3. Construction of a cpDNA Phylogenetic Map
3. Results and Analysis
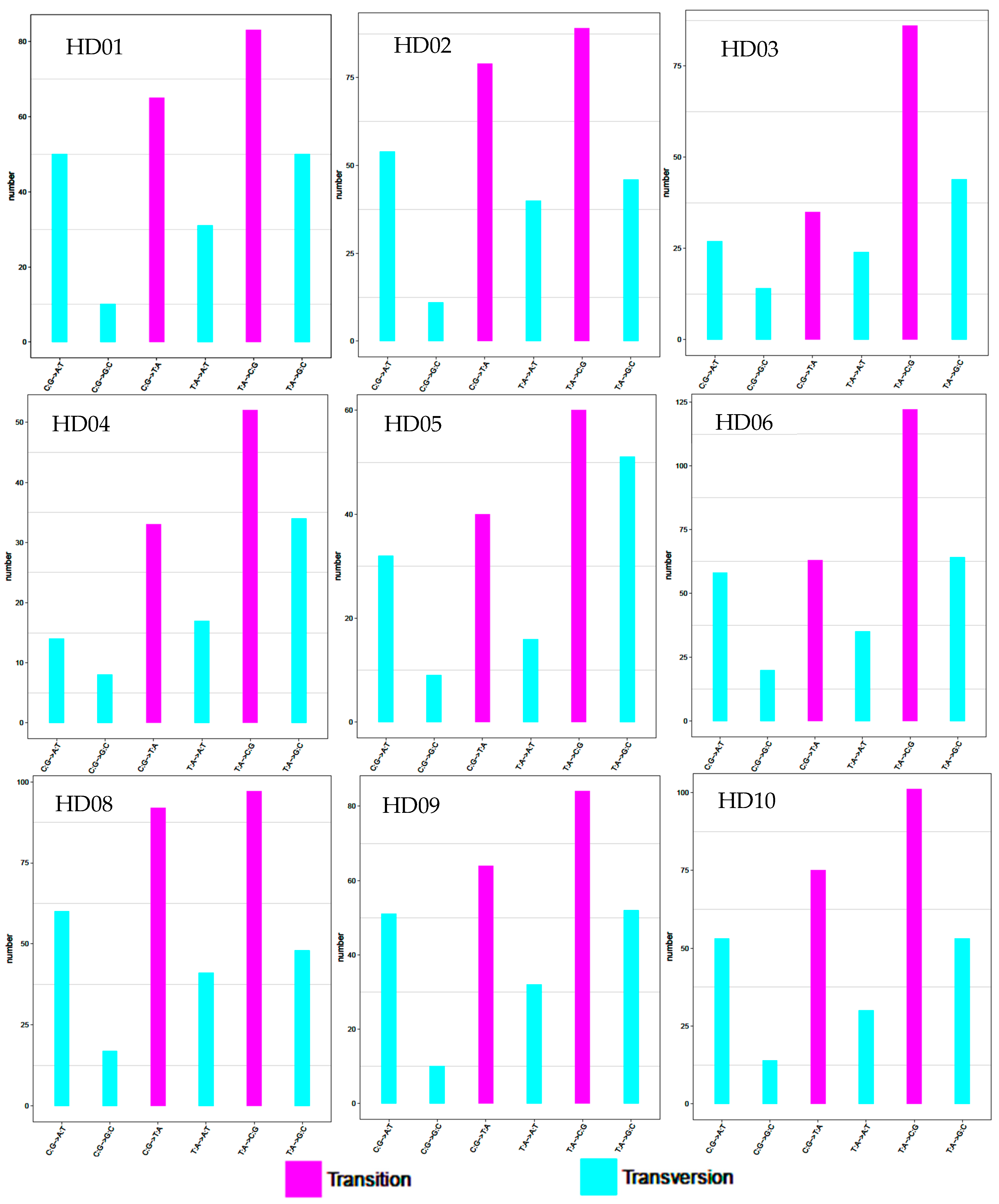
3.1. Identification and Preference Analysis of SNP Mutation Types
3.2. SNP Genotyping
3.3. SNP Annotation
3.4. SNP Comparisons among Samples
3.4.1. Comparisons among the Different Populations of C. gauchowensis and among C. gauchouwensis, C. vietnamensis and the Undetermined Species from Hainan Province
3.4.2. Comparison among C. osmantha, C. gauchowensis and C. vietnamensis
3.4.3. Comparisons among C. gauchowensis, C. vietnamensis, Undetermined Species from Hainan Province and C. osmantha
3.4.4. Comparison between C. giganticarpa and C. meiocarpa
3.4.5. Comparison between C. polyodonta and C. semiserrata
3.4.6. Integrative Comparisons
3.5. Identification of the Kinds of InDels and Calculation of Statistics at the InDel Sites
3.6. InDel Annotation
3.6.1. Exon Regions
3.6.2. Intron Regions
3.6.3. Upstream, Downstream and Intergenic Regions
3.6.4. Example Analysis of InDels
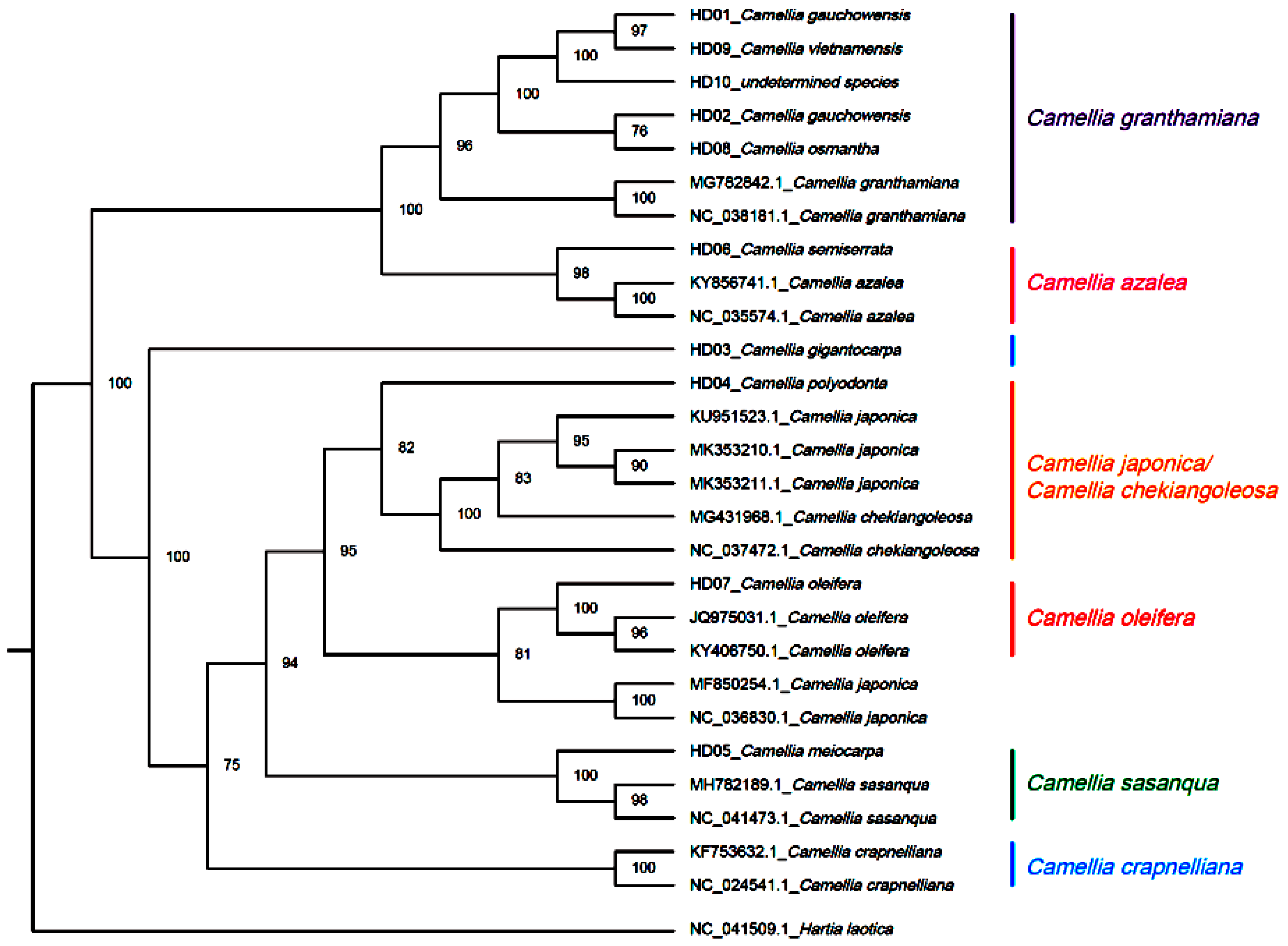
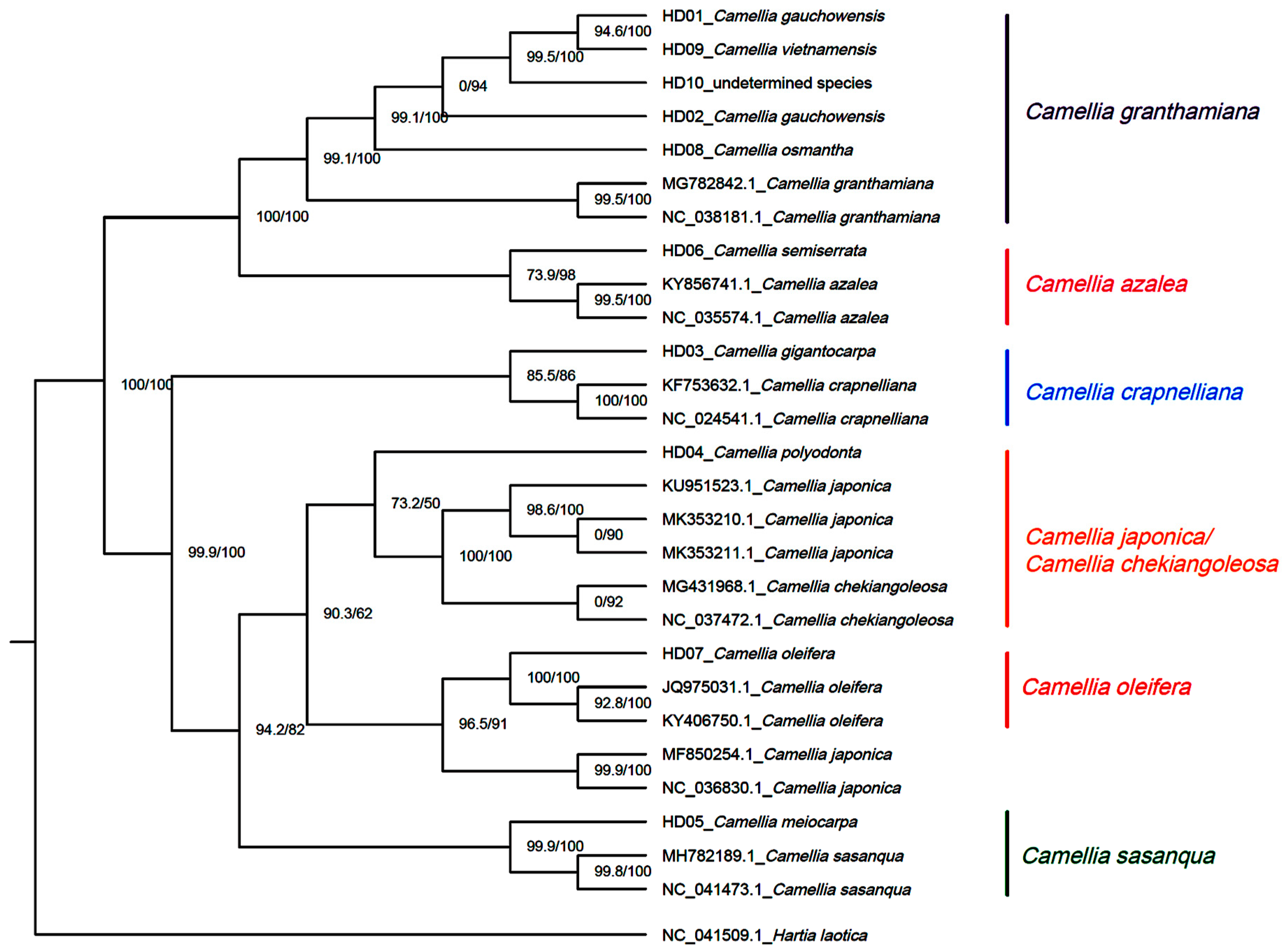
3.7. Phylogenetic Inference
4. Discussion
4.1. Primary Analysis of the Genetic Mechanism Underlying the Phylogenetic Relationships within the Camellia Genus
4.2. Application Value Analysis of SNPs and InDels in the cpDNAs of Oil-Tea Camellia
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- National Forest Farm and Forest Seedling Work Station of State Forestry Administration. Cultivars of Chinese Oil-Tea Camellia; Press of Chinese Forestry: Beijing, China, 2016; pp. 11–15. (In Chinese) [Google Scholar]
- Zhu, B.; Zhong, H.Y.; Cao, Q.M.; Long, Q.Z. Advance in research on biaoactive compounds in Camellia spp. Nonwood For. Res. 2010, 28, 140–145. (In Chinese) [Google Scholar]
- Chen, L.Q.; Yang, W.B.; Li, Y.; Wang, Y.; Fu, D.Q.; Niu, X.Q.; Wang, X.S. The development course and prospect of camellia oil industry in Hainan Island. Mod. Agric. Sci. Technol. 2012, 1, 374. (In Chinese) [Google Scholar]
- Dai, J.; Zhong, S.J. Development Status of Camellia Oil Industry in Hainan and Suggestions. Trop. Agric. Eng. 2017, 41, 61–64. (In Chinese) [Google Scholar]
- Chen, J.; Guo, Y.J.; Hu, X.W.; Zhou, K.B. Comparison of the Chloroplast Genome Sequences of 13 Oil-Tea Camellia Samples and Identification of an Undetermined Oil-Tea Camellia Species From Hainan Province. Front. Plant Sci. 2022, 12, 798581. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Z.; Chen, L.S.; Li, R.F.; Ma, L. Survey and the industry development proposals of Camellia sp. resources in Hainan. Trop. For. 2017, 45, 49–52. (In Chinese) [Google Scholar]
- Ye, Z.C.; Wu, Y.G.; Dai, J.; Zhou, K.B.; Hu, X.W. Distribution of Duckweed Community and the Environmental Factors that Affect the Community Structure. J. Trop. Biol. 2015, 3, 310–314. (In Chinese) [Google Scholar]
- Yuan, J.; Han, Z.Q.; He, S.Y.; Huang, L.Y.; Zhou, N.F. Investigation and Cluster Analysis of Main Morphological and Economical Characters for Oil tea Resource in Hainan Province. J. Plant Genet. Resour. 2014, 15, 1380–1384. (In Chinese) [Google Scholar]
- Hebert, P.D.; Cywinska, A.; Ball, S.L.; De Waard, J.R. Biological identifications through DNA barcodes. Proc. R. Soc. B-Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, M.; Dong, X.; Lin, R.Z.; Fan, J.H.; Chen, Z.D. Evaluation of four commonly used DNA barcoding loci for Chinese medicinal plants of the family Schisandraceae. PLoS ONE 2015, 10, e0125574. [Google Scholar] [CrossRef]
- Li, X.; Yang, Y.; Henry, R.J.; Rossetto, M.; Wang, Y.T.; Chen, S. Plant DNA barcoding: From genes to genome. Biol. Rev. 2015, 90, 157–166. [Google Scholar] [CrossRef]
- Semerikova, S.A.; Semerikov, V.L. Molecular phylogenetic analysis of the genus Abies (Pinaceae) based on the nucleotide sequence of chloroplast DNA. Russ. J. Genet 2014, 50, 7–19. [Google Scholar] [CrossRef]
- Leigh, F.J.; Mackay, I.; Oliveira, H.R.; Gosman, N.E.; Horsnell, R.A.; Jones, H.; White, J.; Powell, W.; Brown, T.A. Using diversity of the chloroplast genome to examine evolutionary history of wheat species. Genet. Resour. Crop Evol. 2013, 60, 1831–1842. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, S.X.; Ji, P.Z.; Gao, L.Z. Phylogeography of Camellia taliensis (Theaceae) inferred from chloroplast and nuclear DNA: Insights into evolutionary history and conservation. BMC Evol. Biol. 2012, 12, 92. [Google Scholar] [CrossRef]
- Zhao, C.L.; Peng, L.Y.; Wang, X.; Chen, J.L.; Wang, L.; Chen, H.; Lai, Z.X.; Liu, S.C. Codon bias and evolution analysis of AtGAI in Amaranthus tricolor L. J. China Agric. Univ. 2019, 24, 10–22. (In Chinese) [Google Scholar]
- Kwak, S.Y.; Lew, T.T.S.; Sweeney, C.J.; Koman, V.B.; Wong, M.H.; Bohmert-Tatarev, K.; Snell, K.D.; Seo, J.S.; Chua, N.H.; Strano, M.S. Chloroplast-selective gene delivery andexpression in planta using chitosan-complexed single-walled carbon nanotubecarriers. Nat. Nanotechnol. 2019, 14, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Song, G.; Wang, N.; Huang, S.; Gao, Y.; Fu, W.; Zhang, M.; Feng, H. A single SNP in Brcer1 results in wax deficiency in Chinese cabbage (Brassica campestris L. ssp. pekinensis). Sci. Hortic. 2021, 282, 110019. [Google Scholar] [CrossRef]
- Ardashir, K. An efficient computational method for screening functional SNPs in plants. J. Theor. Biol. 2010, 265, 55–62. [Google Scholar]
- Kojoma, M.; Seki, H.; Yoshida, S.; Muranaka, T. DNA polymorphisms in the tetrahydrocannabinolic acid (THCA) synthase gene in “drug type” and “fiber-type” Cannabis sativa L. Forensic Sci. Int. 2006, 159, 132–140. [Google Scholar] [CrossRef]
- Miao, H.X.; Sun, P.G.; Zhang, K.X.; Jin, Z.Q.; Xu, B.Y. Research progress on expression regulation mechanism of genes encoding granule-bound starch synthase in plants. Biotechnol. Bull. 2016, 32, 18–23. (In Chinese) [Google Scholar]
- Qi, Y.D.; Zhang, B.G.; Liu, H.T.; Xiao, P.G. Complete chloroplast genome sequences of Schisandra chinensis: Genome struture, comparative analysis, and phylogenetic relationship of basal angiosperms. Sci. China Life Sci. 2017, 7, 728–739. (In Chinese) [Google Scholar]
- Yun, T.; Li, J.M.; Zhou, A.P.; Yan, L.X.; Zong, D.; Li, D.; He, C.Z. Analysis of phylogenetic relationship of populus based on sequence data of chloroplast regions. Plant Physiol. J. Plant Physiol. J. 2015, 51, 1339–1346. (In Chinese) [Google Scholar]
- Chen, C.; Ma, C.; Ma, J.; Liu, S.; Chen, L. Sequencing of Chloroplast Genome of Camellia sinensis and Genetic Relationship for Camellia Plants Based on Chloroplast DNA Sequences. J. Tea Sci. 2014, 34, 371–380. (In Chinese) [Google Scholar]
- Liang, G.X.; Liu, K.; Ma, J.L.; Chen, G.C.; Ye, H.; Jiang, Z.P. Molecular taxonomy and identification of Camellia osmantha. Nonwood For. Res. 2017, 35, 26–29. (In Chinese) [Google Scholar]
- Wang, D.X.; Ye, H.; Ma, J.L.; Zhou, Z.D. Evaluation and selection of Camellia osmantha germplasm resources. Nonwood For. Res. 2014, 32, 159–162. (In Chinese) [Google Scholar]
- Luo, C.Q.; Tan, X.F.; Qi, L.L. A classification summary on plant of Genus Camellia. J. Cent. South For. Coll. 1999, 19, 78–81. (In Chinese) [Google Scholar]
- Stahl, P.; Mirom, Y.L.; Stern, R.A.; Goldway, M. Comparing ‘Iriet’ and ‘Ettinger’ avocado cultivars as pollinators of ‘Hass’ using SNPs for paternal identification. Sci. Hortic. 2019, 248, 50–57. [Google Scholar] [CrossRef]
- Chagné, D.; Gasic, K.; Crowhurst, R.N.; Han, Y.; Bassett, H.C.; Bowatte, D.R.; Lawrence, T.J.; Rikkerink, E.H.A.; Gardiner, S.E.; Korban, S.S. Development of a set of SNP markers present in expressed genes of the apple. Genomics 2008, 92, 353–358. [Google Scholar] [CrossRef]
- Dong, Q.H.; Cao, X.; Yang, G.; Yu, H.P.; Nicholas, K.K.; Wang, C.; Fang, J.G. Discovery and characterization of SNPs in Vitis vinifera and genetic assessment of some grapevine cultivars. Sci. Hortic. 2010, 125, 233–238. [Google Scholar] [CrossRef]
- Bielsa, B.; Jiwan, D.; i Marti, A.F.; Dhingra, A.; Rubio-Cabetas, M.J. Detection of SNP and validation of a SFP InDel (deletion) in inverted repeat region of the Prunus species chloroplast genome. Sci. Hortic. 2014, 168, 108–112. [Google Scholar] [CrossRef]
- Osorio, C.E.; Udall, J.A.; Salvo-Garrido, H.; Maureira-Butler, I.J. Development and characterization of InDel markers for Lupinus luteus L. (Fabaceae) and cross-species amplification in other Lupin species. Electron. J. Biotechnol. 2018, 31, 44–47. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Samples | HD01 | HD02 | HD03 | HD04 | HD05 | HD06 | HD08 | HD09 | HD10 |
|---|---|---|---|---|---|---|---|---|---|
| Homozygous SNP | 143 | 152 | 80 | 57 | 77 | 129 | 156 | 142 | 128 |
| Heterozygous SNP | 3 | 15 | 70 | 44 | 54 | 104 | 43 | 9 | 70 |
| Total | 146 | 167 | 150 | 101 | 131 | 233 | 199 | 151 | 198 |
| Samples | Regions of Gene | Gene (Numbers of the SNPs) | Total |
|---|---|---|---|
| HD01 | Downstream | psbA(3), rps16(1), psbI(15), atpA(2), atpI(2), petN(4), ndhJ(5) | 32 |
| Exon | psbA(1), Matk(2), psbK(2), rps2(1), rpoC2(5), rpoB(5), psaA(1), ndhJ(1), atpB(3), rbcL(1), accD(1), accD(3), ycf4(2) | 28 | |
| Intergenic space | rpoB(1), psbM(2), rpl20(43) | 46 | |
| Intron | rps16(2), atpF(1), rpoC1(2), ycf3(1) | 6 | |
| Upstream | rps16(3), psbK(1), psbI(2), rpoB(3), psbM(1), psbD(1), rps4(1), ndhC(2), atpB(2), rpl20(3) | 19 | |
| Upstream; downstream | pabA;matK(1)/psbI; psbK(5)/atpH; atpI(2)/psaI; accD(1)/cemA; ycf4(2)/petG; petL(1)/psaJ; petG(1)/psaJ(1)/rpl20; rps18(1) | 15 | |
| Total | 146 | ||
| HD02 | Downstream | psbA(3), rps16(1), psbI(15), atpA(2), atpI(2), petN(3), ndhJ(6), psaB(2), rpl20(1) | 35 |
| Exon | matk(1), psbK(2), rps2(1), rpoC2(3), rpoC2(1), rpoB(4), rpoB(2), psaA(1), ndhJ(1), ndhJ(3), atpE(1), atpE(3), atpB(3), rbcL(1),accD(1), accD(1), accD(2), ycf3(1), psbF(1), psbF(1), psbF(1) | 34 | |
| Intergenic space | psbI(1), psbM(2), rpl20(52) | 55 | |
| Intron | rps16(2), atpF(1), rpoC1(2), ycf3(1) | 6 | |
| Upstream | rps16(2), psbK(1), psbI(2), rpoB(5), petN(1), psbM(2), rps4(1), ndhC(2), atpB(2), petG(1), rpl20(3) | 22 | |
| Upstream; downstream | pabA; matK(1)/psbI; psbK(4)/atpA; atpF(1)/atpH; atpI(2)/psaI; accD(1)/cemA; ycf4(2)/petG; petL(1)/psaJ; petG(1)/psaJ(1)/rpl20; rps18(1) | 15 | |
| Total | 167 | ||
| HD13, HD10 –HD12 | Downstream | psbA(3), rps16(1), psbI(12), atpA(2), atpI(2), petN(4), ndhJ(5) | 29 |
| Exon | psbA(1), matk(2), psbK(2), rps2(1), rpoC2(4), rpoC2(1), rpoB(3), rpoB(2), psbC(1), psbC(4), psaA(1), ndhJ(2), ndhJ(6), atpE(1), atpE(4), atpB(3), rbcL(1), accD(1), accD(1), accD(1), ycf4(1), ycf4(1), psbJ(2), psbJ(1), psbL(3), psbF(5), psbF(1), psbE(3), psbE(1), petL(4), petL(1), petG(1) | 66 | |
| Intergenic space | rpoB(1), psbM(2), rpl20(57) | 60 | |
| Intron | rps16(2), atpF(1), rpoC1(2), ycf3(1) | 6 | |
| Upstream | rps16(3), psbK(1), psbI(2), rpoB(3), psbM(4), psbD(1), rps4(1), ndhC(1), atpB(1), rpl20(3) | 20 | |
| Upstream; downstream | pabA;matK(1)/psbI; psbK(5)/atpH; atpI(2)/psaI; accD(1)/cemA; ycf4(2)/psaJ; petG(4)/psaJ(1)/rpl20; rps18(1) | 17 | |
| Total | 198 | ||
| HD09 | Downstream | psbA(3), rps16(1), psbI(15), atpA(2), atpI(2), petN(4), ndhJ(5) | 32 |
| Exon | psbA(1), Matk(2), psbK(2), rps2(1), rpoC2(4), rpoC2(1), rpoB(3), rpoB(2), psaA(1), ndhJ(2), atpB(3), rbcL(1), accD(1), accD(1), accD(2), ycf4(1), ycf4(1) | 29 | |
| Intergenic space | rpoB(1), psbM(2), rpl20(49) | 52 | |
| Intron | rps16(2), atpF(1), rpoC1(2), ycf3(1) | 6 | |
| Upstream | rps16(3), psbK(1), psbI(2), rpoB(3), psbM(1), psbD(1), rps4(1), ndhC(1), atpB(1), rpl20(3) | 17 | |
| Upstream; downstream | pabA;matK(1)/psbI; psbK(5)/atpH; atpI(2)/psaI;accD(1)/cemA; ycf4(2)/petG; petL(1)/psaJ; petG(1)/psaJ(1)/rpl20; rps18(1) | 15 | |
| Total | 151 | ||
| HD08 | Downstream | psbA(3), rps16(2), psbI(20), atpA(3), atpI(2), petN(7), ndhJ(5), petA(1) | 43 |
| Exon | matk(1), psbK(2), rps2(1), rpoC2(3), rpoC2(2), rpoB(3), rpoB(2), psaA(1), psaA(2), ndhJ(2), atpE(4), atpE(5), atpB(3), rbcL(1), accD(1), accD(1), accD(2), ycf4(1), psbF(1), psbF(1), petG(1), rpl20(1) | 40 | |
| Intergenic space | psbM(4), rps4(1), rpl20(60) | 65 | |
| Intron | rps16(3), atpF(1), rpoC1(3), ycf3(3) | 10 | |
| Upstream | rps16(3), psbK(1), psbI(2), rpoB(3), psbM(2), rps4(1), ndhC(1), atpB(2), rpl20(5) | 20 | |
| Upstream; downstream | pabA; matK(1)/psbI; psbK(4)/atpH; atpI(2)/rpoC2; rpoC1(1) /ndhJ; ndhC(3)/psaI; accD(1)/cemA; ycf4(2)/petA; cemA(2)/petG; petL(1)/psaJ; petG(1)/psaJ(1)/rpl33(1)/rpl20; rps18(1) | 21 | |
| Total | 199 | ||
| HD03 | Downstream | psbA(1), psbI(12), atpA(1), atpI(1), petN(7), petA(3) | 25 |
| Exon | matk(1), psbK(1), atpF(1), rps2(1), rpoC2(1), rpoC1(1), rpoB(2), psbC(1), psbC(3), ndhJ(1), ndhJ(2), ndhJ(8), atpE(3), atpE(5), atpB(2), atpB(1), rbcL(1), accD(2), ycf4(1), psbJ(3), psbJ(2), psbL(3), psbF(1), psbF(1), petL(4), petL(2), petG(2) | 56 | |
| Intergenic space | rps4(2), psbM(2), rpl20(31) | 35 | |
| Intron | rps16(2), atpF(2), ycf3(2) | 6 | |
| Upstream | rps16(1), psbK(1), psbI(2), rpoB(1), psbM(1), ndhC(1), rpl20(1) | 8 | |
| Upstream; downstream | psbI; psbK(1)/ndhJ; ndhC(2)/psaI; accD(1)/psbJ; psbE(2)/psaJ; petG(13)/psaJ(1) | 20 | |
| Total | 150 | ||
| HD04 | Downstream | psbA(1), psbI(12), atpI(1), petN(2), atpB(1) | 17 |
| Exon | psbK(1), rpoC1(1), rpoB(3), rpoB(1), ndhJ(4), ndhJ(1), ndhJ(7), atpE(2), atpE(6), atpB(2), ycf4(1), psbJ(3), psbJ(2), psbL(3), psbF(1), psbF(1), psbF(1) | 40 | |
| Intergenic space | psbM(1), rpl20(19) | 20 | |
| Intron | rps16(2), atpF(1), ycf3(1) | 4 | |
| Upstream | rps16(1), psbI(1), atpF(1), rpoB(1), psbM(5), rps4(1), petG(1), rpl20(1) | 12 | |
| Upstream; downstream | psbI; psbK(1)/ndhJ; ndhC(2)/ndhC; atpE(1)/psaJ; accD(1)/psaJ; petG(2)/psaJ(1) | 8 | |
| Total | 101 | ||
| HD05 | Downstream | psbA(1), psbI(12), atpI(1), petN(2), atpB(1) | 17 |
| Exon | matK(2), atpF(1), rps2(1), rpoC2(2), rpoB(2), rpoB(1), psbC(2), psaB(1), ndhJ(4), ndhJ(6), atpE(1), atpE(4), atpB(1), ycf4(1), psbJ(2), psbJ(1), psbL(3), psbF(1), psbF(1), psbF(1), petL(4), petL(2), petG(2) | 47 | |
| Intergenic space | psbI(1), psbM(1), rpl20(39) | 41 | |
| Intron | rps16(2), atpF(1), ycf3(1) | 4 | |
| Upstream | rps16(1), psbK(1), psbI(2), rpoB(1), ndhC(3), atpB(1), rpl20(1) | 10 | |
| Upstream; downstream | psbA; matk(1)/psbI; psbK(2)/atpF(2)/psaI; accD(1)/psaJ; petG(5)/psaJ(1) | 12 | |
| Total | 131 | ||
| HD06 | Downstream | psbA(3), psbI(15), atpA(2), atpI(1), petN(7), ndhJ(4) | 32 |
| Exon | matK(1), psbK(2), atpH(1), rps20(1), rpoC2(1), rpoC2(1), rpoB(3), rpoB(3), psbC(1), psbC(4), ndhJ(1), ndhJ(4), ndhJ(7), atpE(6), atpE(9), atpB(5), atpB(6), rbcL(1), accD(1), accD(1), ycf4(1), psbJ(2), psbJ(1), psbL(1), psbF(1), psbF(1), psbF(1), psbE(2), psbE(1), petL(4), petL(1), petL(2), petG(2), rps18(1) | 80 | |
| Intergenic space | psbM(3), rpl20(50) | 53 | |
| Intron | rps16(2), atpF(1), rpoC1(1), ycf3(1) | 5 | |
| Upstream | matK(1), rps16(3), psbK(1), psbI(2), rpoB(3), psbM(6), rps4(1), ndhC(1), atpB(2), petG(1), rpl20(2) | 23 | |
| Upstream; downstream | psbI; psbK(4)/atpH; atpI(2)/psaA; ycf3(1)/ndhJ; ndhC(2)/psaJ; accD(1)/cemA; ycf4(2)/psaI(1)/petG; petL(4)/petG(1)/psaJ; petG(20)/psaJ(1)/rpl20; rps18(1) | 40 | |
| Total | 233 | ||
| Samples | HD01 | HD02 | HD03 | HD04 | HD05 | HD06 | HD08 | HD09 | HD10 |
|---|---|---|---|---|---|---|---|---|---|
| Total | 28 | 34 | 56 | 40 | 47 | 80 | 40 | 29 | 66 |
| Nonsynonymous SNV | 11 | 12 | 27 | 18 | 21 | 32 | 16 | 11 | 31 |
| Stopgain SNV | 1 | 2 | 2 | 2 | 1 | 2 | 1 | 1 | 2 |
| Stopiloss SNV | - | - | - | - | - | 1 | - | - | 1 |
| Synonymous SNV | 16 | 20 | 27 | 20 | 25 | 45 | 23 | 17 | 32 |
| Regions of Gene | Gene (Numbers of the SNPs) | Total | |
|---|---|---|---|
| HD01 HD02 HD13 | Downstream | psbA(3), rps16(1), atpA(2), atpI(2) | 18 |
| Exon | matk(2), rps2(1), psaA(1), atpB(3), rbcL(1), accD(1) | 9 | |
| Intergenic space | rpoB(1), psbM(2) | 3 | |
| Intron | rps16(2), atpF(1), rpoC1(2), ycf3(1) | 6 | |
| Upstream | psbK(1), psbI(2), rps4(1), rpl20(3) | 7 | |
| Upstream; downstream | pabA; matK(1)/atpH; atpI(2)/psaI; accD(1)/cemA; ycf4(2)/psaJ(1)/rpl20; rps18(1) | 8 | |
| Total | 51 | ||
| HD01 HD09 HD10– HD13 | Downstream | psbA(3), rps16(1), atpA(2), atpI(2), petN(4), ndhJ(5) | 17 |
| Exon | psbA(1), matk(2), psbK(2), rps2(1), rpoC2(5), rpoB(5), atpB(3), rbcL(1), accD(1), ycf4(2) | 23 | |
| Intergenic space | rpoB(1), psbM(2) | 3 | |
| Intron | rps16(2), atpF(1), rpoC1(2), ycf3(1) | 6 | |
| Upstream | rps16(3), psbK(1), psbI(2), rpoB(3), psbD(1), rps4(1), rpl20(3) | 14 | |
| Upstream; downstream | pabA; matK(1)/psbI; psbK(5)/atpH; atpI(2)/psaI; accD(1)/cemA; ycf4(2)/psaJ(1)/rpl20; rps18(1) | 13 | |
| Total | 76 | ||
| HD01 HD08 HD09 | Downstream | psbA(3), atpI(2), ndhJ(5) | 10 |
| Exon | rps2(1), rpoC2(5), rpoB(5), atpB(3), rbcL(1), accD(1), ycf4(2) | 18 | |
| Intron | atpF(1), ycf3(1) | 2 | |
| Upstream | rps16(3), psbK(1), psbI(2), rpoB(3), rps4(1), atpB(1) | 11 | |
| Upstream; downstream | psaI; accD(1)/cemA; ycf4(2)/petG; petL(1)/psaJ; petG(1)/psaJ(1)/rpl20; rps18(1) | 7 | |
| Total | 48 | ||
| HD01 HD02 HD08 HD09 HD10– HD13 | Downstream | psbA(3), atpI(2) | 5 |
| Exon | rps2(1), atpB(3), rbcL(1), accD(1) | 6 | |
| Intron | atpF(1), ycf3(1) | 2 | |
| Upstream | psbK(1), psbI(2), rps4(1) | 4 | |
| Upstream; downstream | cemA; ycf4(2)/psaJ(1)/rpl20; rps18(1) | 4 | |
| Total | 21 | ||
| HD03 HD05 | Downstream | psbA(1), psbI(12), atpA(1) | 14 |
| Exon | matk(1), atpF(1), rps2(1), ycf4(1), petL(6), petG(2), psbF(1), psbF(1) | 14 | |
| Intron | rps16(2) | 2 | |
| Upstream | rps16(1), psbK(1), psbI(2), rpoB(1), rpl20(1) | 6 | |
| Upstream; downstream | psaI; accD(1)/psaJ(1) | 2 | |
| Total | 38 | ||
| HD04 HD06 | Downstream | atpI(1) | 1 |
| Exon | rpoB(3), ndhJ(1), ndhJ(4), ndhJ(7), psbF(1), psbF(1), psbF(1) | 18 | |
| Intron | rps16(2), atpF(1), ycf3(1) | 4 | |
| Upstream | rps4(1), petG(1) | 2 | |
| Upstream; downstream | psaJ; accD(1)/psaJ(1) | 2 | |
| Total | 27 | ||
| Intron | rps16(2) | 2 | |
| Upstream; downstream | accD(1)/psaJ(1) | 1 | |
| Total | 3 | ||
| All samples | Total | 0 | |
| Samples | HD01 | HD02 | HD03 | HD04 | HD05 | HD06 | HD08 | HD09 | HD10 |
|---|---|---|---|---|---|---|---|---|---|
| Deletion InDels | 22 | 28 | 25 | 13 | 17 | 22 | 25 | 21 | 20 |
| Insertion InDels | 17 | 17 | 13 | 8 | 7 | 16 | 18 | 17 | 16 |
| Total | 39 | 45 | 38 | 21 | 24 | 38 | 43 | 38 | 36 |
| The ratio of deletion to insertion | 1.29 | 1.65 | 1.92 | 1.63 | 2.43 | 1.38 | 1.39 | 1.24 | 1.25 |
| Samples | Regions of Gene | Gene (Numbers of the SNPs) | Total |
|---|---|---|---|
| HD01 | Downstream | psbA(1), rps16(1), psbI(3), atpA(2), psbZ(1), psaB(1), petA(1) | 10 |
| Intergenic space | psbM(1), rps4(1), rpl20(4) | 6 | |
| Intron | rps16(2), ycf3(1) | 3 | |
| Upstream | matK(2), psbK(1), psbD(1), ycf3(1), rps4(1), ndhC(1) | 7 | |
| Upstream; downstream | psbI; psbK(1)/atpI(1)/psaI; accD(1)/psaI(1)/ycf4; accD(1)/cemA; ycf4(3)/petA; cemA(1)/psaJ(1)/rpl33(1)/rpl20; rps18(2) | 13 | |
| Total | 39 | ||
| HD02 | Downstream | psbA(1), rps16(1), psbI(3), atpA(4), psbZ(1), psaB(3), petA(2) | 15 |
| Intergenic space | psbM(1), rps4(1), rpl20(6) | 8 | |
| Intron | rps16(2), ycf3(1) | 3 | |
| Upstream | matK(2), rps16(1), psbK(1), psbD(1), ycf3(1), rps4(1), ndhC(1) | 8 | |
| Upstream; downstream | psbA; matK(1)/psbI; psbK(1)/atpI(1)/ndhJ; ndhC(1)/psaI; accD(1)/ycf4; accD(1)/cemA; ycf4(1)/petA; cemA(1)/psaJ(1)/rpl33(1)/rpl20; rps18(1) | 11 | |
| Total | 45 | ||
| HD03 | Downstream | psbA(1), psbI(2), atpA(3), atpI(1), psaB(1), ndhJ(1), petA(3) | 12 |
| Exon | rpoC2(1) | 1 | |
| Intergenic space | psbM (2), rps4(1), rpl20(4) | 7 | |
| Upstream | matK(1), rps16(2), psbK(1), yfc3(1), rps4(2), ndhC(2), petG(1) | 10 | |
| Upstream; downstream | psbI; psbK(1)/psaI; accD(1)/cemA; ycf4(2)/petA; cemA(1)/psaJ(1)/rpl33(1)/ rpl20; rps18(1) | 8 | |
| Total | 38 | ||
| HD04 | Downstream | psbI(2), atpA(3), psaB(1) | 6 |
| Intergenic space | rpl20(3) | 3 | |
| Upstream | ndhC(2) | 2 | |
| Upstream; downstream | atpI(1)/psaA; ycf3(1)/ycf3; rps4(1)/psaI; accD(1)/cemA; ycf4(2)/petA; cemA(1)/psaJ(1)/rpl33(1)/rpl20; rps18(1) | 10 | |
| Total | 21 | ||
| HD05 | Downstream | psbA(1), psbI(2), atpA(3), psaB(1) | 7 |
| Intergenic space | rpl20(5) | 5 | |
| Upstream | rps16(2), ndhC(1) | 3 | |
| Upstream; downstream | psbI; psbK(1)/atpI; rps2(1)/psaI; accD(1)/cemA; ycf4(2)/petA; cemA(1)/psaJ(1)/rpl33(1)/rpl20; rps18(1) | 9 | |
| Total | 24 | ||
| HD06 | Downstream | psbA(1), psbI(3), atpA(2), psbZ(1), psaB(2), petA(1) | 10 |
| Intergenic space | psbM(1), rps4(2), rpl20(7) | 10 | |
| Intron | rps16(2), ycf3(1) | 3 | |
| Upstream | matK(4), ycf3(1), ndhC(2) | 7 | |
| Upstream; downstream | psaI; accD(1)/ycf4; accD(1)/cemA; ycf4(2)/petA; cemA(1)/psaJ(1)/rpl33(1)/ rpl20; rps18(1) | 8 | |
| Total | 38 | ||
| HD08 | Downstream | psbA(1), rps16(2), psbI(3), atpA(2), psbZ(1), psaB(2), petA(2) | 13 |
| Intergenic space | psbM(1), rps4(1), rpl20(7) | 9 | |
| Intron | rps16(2), ycf3(1) | 3 | |
| Upstream | matK(2), rps16(1), psbK(1), petN(1), psbM(1), psbD(1), ycf3(1), ndhC(1) | 9 | |
| Upstream; downstream | psbI; psbK(1)/atpH; atpI(1)/atpI(1)/psaI; accD(1)/ycf4; accD(1)/cemA; ycf4(2)/petA; cemA(1)/rpl33(1)/rpl20; rps18(1) | 9 | |
| Total | 43 | ||
| HD09 | Downstream | psbA(1), rps16(1), atpI(3), atpA(1), psbZ(1), psaB(1), petA(1) | 9 |
| Intergenic space | psbM (1), rps4(1), rpl20(4) | 6 | |
| Intron | rps16(2), ycf3(1) | 3 | |
| Upstream | matK(2), psbK(1), psbD(1), ycf3(1), rps4(1), ndhC(1) | 7 | |
| Upstream; downstream | psbI; psbK(1)/atpI(1)/psaI; accD(1)/psaI(1)/ycf4; accD(1)/cemA; ycf4(3)/petA; cemA(1)/psaJ(1)/rpl33(1)/rpl20; rps18(2) | 13 | |
| Total | 38 | ||
| HD10 | Downstream | psbI(1), rps16(1), psbI(1), atpA(2), psbZ(1), psaB(1), petA(1) | 8 |
| Intergenic space | psbZ(1), rps4(1), rpl20(4) | 6 | |
| Intron | rps16(1), ycf3(1) | 2 | |
| Upstream | matK(2), psbK(1), psbD(1), ycf3(1), rps4(1), ndhC(1) | 7 | |
| Upstream; downstream | psbI; psbK(1)/atpI(1)/psaI; accD(1)/psaI(1)/ycf4; accD(1)/cemA; ycf4(3)/petA; cemA(1)/psaJ(1)/rpl33(1)/rpl20; rps18(2) | 13 | |
| Total | 36 | ||
| Samples | InDels (Insertion/Deletion) | Total | Samples | InDels (Insertion/Deletion) | Total |
| HD01 HD09– HD13 | TATAATA (Deletion) | 4 | HD05 | A (Insertion) | 5 |
| AATAT (Deletion) | TATAATA (Deletion) | ||||
| T (Deletion) | CTTAC (Deletion) | ||||
| T (Deletion) | T (Deletion) | ||||
| TCTTTT (Deletion) | |||||
| HD02 | TATAATA (Deletion) | 6 | HD06 | TATAATA (Deletion) | 7 |
| TCATGAT (Deletion) | AATAT (Deletion) | ||||
| AT (Insertion) | AT (Insertion) | ||||
| AATAT (Deletion) | T (Deletion) | ||||
| T (Deletion) | T (Insertion) | ||||
| T (Deletion) | TATGGTAATCCA (Deletion) | ||||
| T (Deletion) | |||||
| HD03 | TATAATA (Deletion) | 4 | HD08 | TATAATA (Deletion) | 7 |
| A (Insertion) | T (Insertion) | ||||
| A (Deletion) | AATAT (Deletion) | ||||
| T (Deletion) | T (Deletion) | ||||
| HD04 | TATAATA (Deletion) | 3 | T (Insertion) | ||
| AATAG (Insertion) | TATGGTAATCCA (Deletion) | ||||
| T (Deletion) | T (Deletion) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Zhou, K.; Hu, X. Comparisons of Chloroplast Genome Mutations among 13 Samples of Oil-Tea Camellia from South China. Genes 2023, 14, 1083. https://doi.org/10.3390/genes14051083
Chen J, Zhou K, Hu X. Comparisons of Chloroplast Genome Mutations among 13 Samples of Oil-Tea Camellia from South China. Genes. 2023; 14(5):1083. https://doi.org/10.3390/genes14051083
Chicago/Turabian StyleChen, Jing, Kaibing Zhou, and Xinwen Hu. 2023. "Comparisons of Chloroplast Genome Mutations among 13 Samples of Oil-Tea Camellia from South China" Genes 14, no. 5: 1083. https://doi.org/10.3390/genes14051083