
Systematic Large Fragment Deletions in the Genome of Synechococcus elongatus and the Consequent Changes in Transcriptomic Profiles
 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Strains and Culture Conditions
2.2. Construction of CRISPR/Cpf1-Based Genome Editing Plasmids
2.3. Generation of Genome Deletions in Synechococcus 7942
2.4. RNA Extraction
2.5. Library Preparation and Illumina Hiseq Sequencing
2.6. Differential Expression Analysis and Functional Enrichment
2.7. Graphic Plotting of RNA-Seq
3. Results
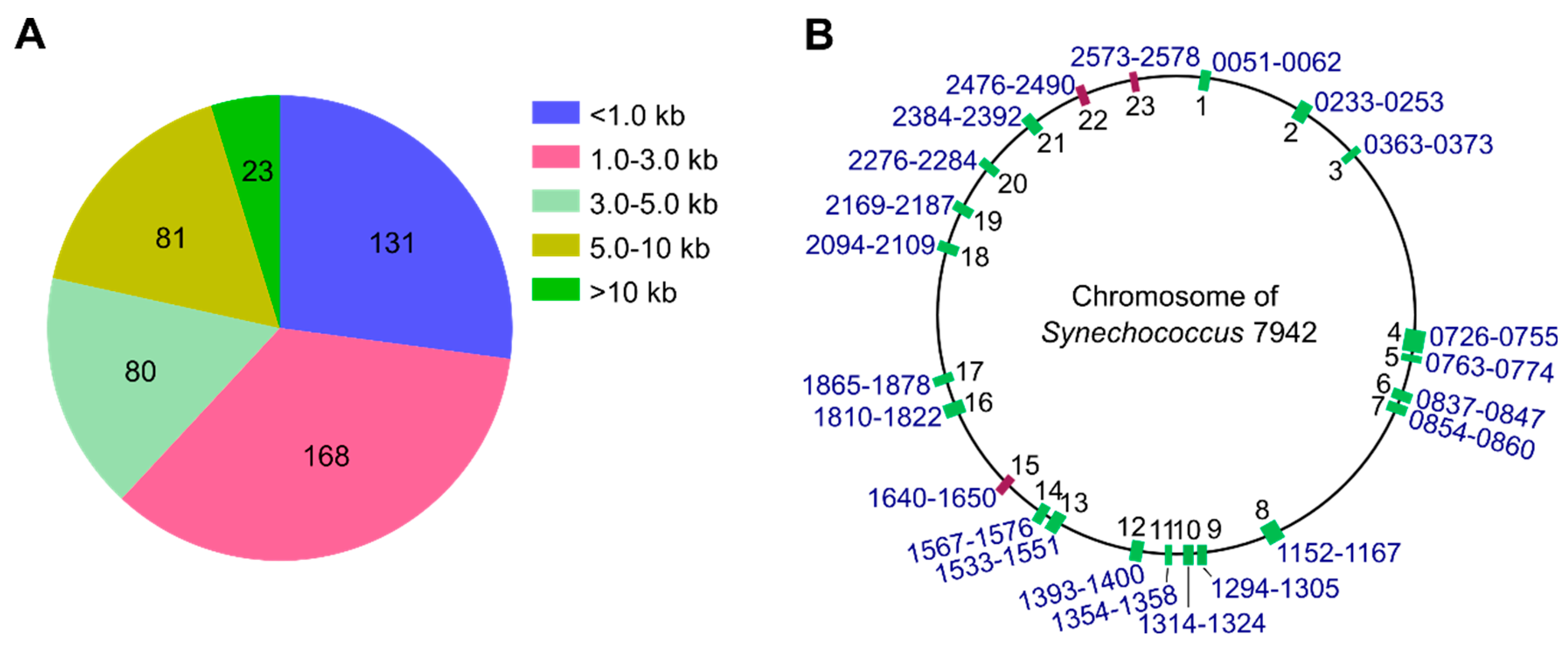
3.1. Identification of over 10 kb Dispensable Chromosomal Regions in Synechococcus 7942
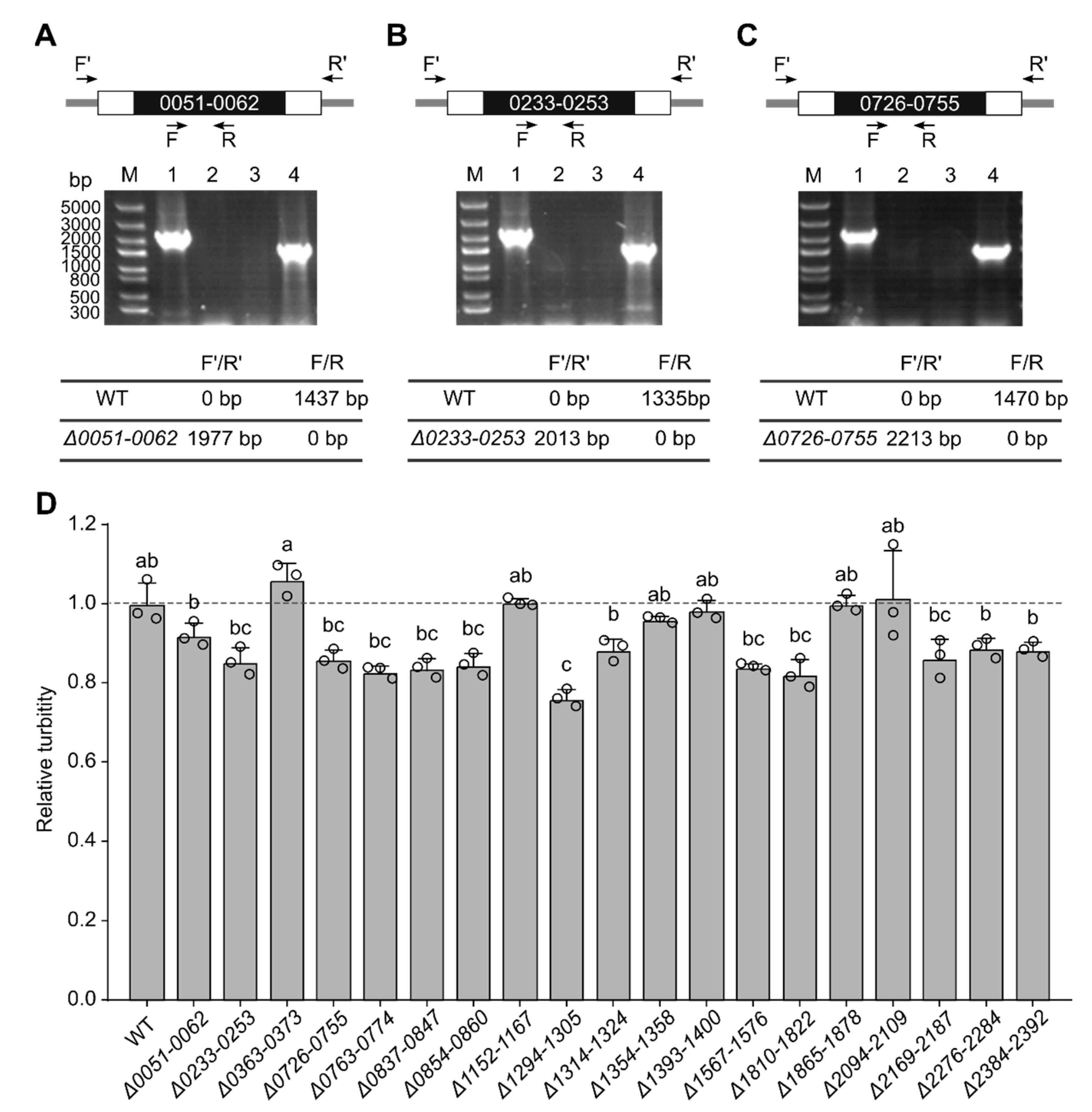
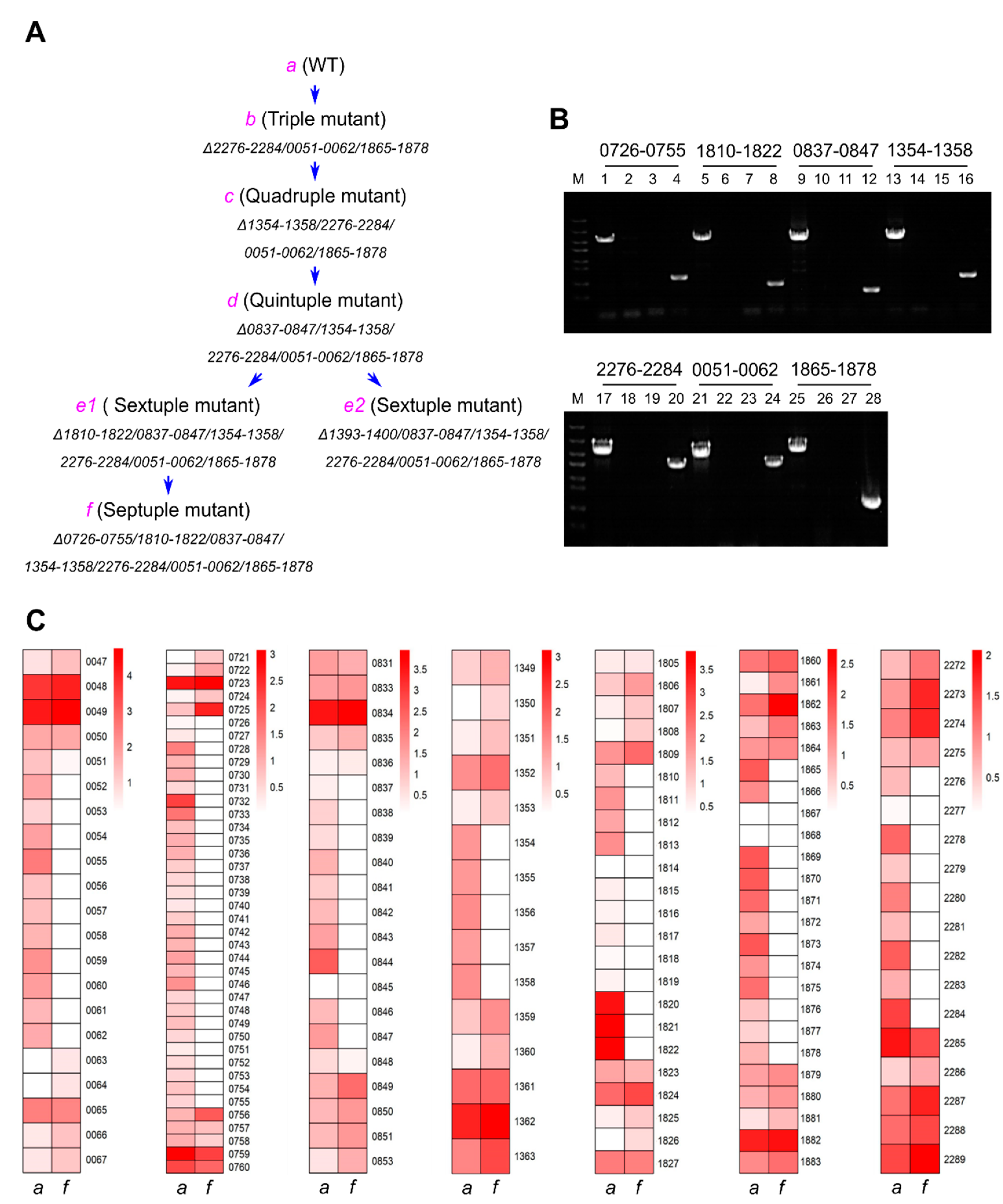
3.2. Systematic Large Fragment Deletions
3.3. Large-Scale Gene Upregulation in Response to Genome Reduction
3.4. Growth of Genome-Reduced Strains under Different Light and CO2 Conditions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- LeBlanc, N.; Charles, T. Bacterial genome reductions: Tools, applications, and challenges. Front. Genome Ed. 2022, 4, 957289. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Yang, Q.; Geng, B.; Jiang, S.; Yang, S.; Li, X.; Cai, Y.; Dai, J. Whole genome engineering by synthesis. Sci. China Life Sci. 2018, 61, 1515–1527. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, M.; Ying, B.-W. Experimental challenges for reduced genomes: The cell model Escherichia coli. Microorganisms 2019, 8, 3. [Google Scholar] [CrossRef] [Green Version]
- Pál, C.; Papp, B.; Pósfai, G. The dawn of evolutionary genome engineering. Nat. Rev. Genet. 2014, 15, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Knoot, C.J.; Ungerer, J.; Wangikar, P.P.; Pakrasi, H.B. Cyanobacteria: Promising biocatalysts for sustainable chemical production. J. Biol. Chem. 2018, 293, 5044–5052. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Sung, B.H.; Kim, M.S.; Blattner, F.R.; Yoon, B.H.; Kim, J.H.; Kim, S.C. Metabolic engineering of a reduced-genome strain of Escherichia coli for L-threonine production. Microb. Cell Fact. 2009, 8, 2. [Google Scholar] [CrossRef] [Green Version]
- Morimoto, T.; Kadoya, R.; Endo, K.; Tohata, M.; Sawada, K.; Liu, S.; Ozawa, T.; Kodama, T.; Kakeshita, H.; Kageyama, Y. Enhanced recombinant protein productivity by genome reduction in Bacillus subtilis. DNA Res. 2008, 15, 73–81. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Uchiyama, T.; Ōmura, S.; Cane, D.E.; Ikeda, H. Genome-minimized Streptomyces host for the heterologous expression of secondary metabolism. Proc. Natl. Acad. Sci. USA 2010, 107, 2646–2651. [Google Scholar] [CrossRef] [Green Version]
- Baumgart, M.; Unthan, S.; Kloß, R.; Radek, A.; Polen, T.; Tenhaef, N.; Müller, M.F.; Küberl, A.; Siebert, D.; Brühl, N.; et al. Crynebacterium glutamicum chassis C1*: Building and testing a novel platform host for synthetic biology and industrial biotechnology. ACS Synth. Biol. 2018, 7, 132–144. [Google Scholar] [CrossRef]
- Knott, G.J.; Doudna, J.A. CRISPR-Cas guides the future of genetic engineering. Science 2018, 361, 866–869. [Google Scholar] [CrossRef] [Green Version]
- Ungerer, J.; Pakrasi, H.B. Cpf1 Is A Versatile Tool for CRISPR Genome Editing Across Diverse Species of Cyanobacteria. Sci. Rep. 2016, 6, 39681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wendt, K.E.; Ungerer, J.; Cobb, R.E.; Zhao, H.; Pakrasi, H.B. CRISPR/Cas9 mediated targeted mutagenesis of the fast growing cyanobacterium Synechococcus elongatus UTEX 2973. Microb. Cell Fact. 2016, 15, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; Van Der Oost, J.; Regev, A. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonfara, I.; Richter, H.; Bratovic, M.; Le Rhun, A.; Charpentier, E. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 2016, 532, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Niu, T.C.; Lin, G.M.; Xie, L.R.; Wang, Z.Q.; Xing, W.Y.; Zhang, J.Y.; Zhang, C.C. Expanding the potential of CRISPR-Cpf1-based genome editing technology in the cyanobacterium Anabaena PCC 7120. ACS Synth. Biol. 2019, 8, 170–180. [Google Scholar] [CrossRef]
- Choubeh, R.R.; Wientjes, E.; Struik, P.C.; Kirilovsky, D.; Van Amerongen, H. State transitions in the cyanobacterium Synechococcus elongatus 7942 involve reversible quenching of the photosystem II core. Biochim. Biophys. Acta (BBA) Bioenerg. 2018, 1859, 1059–1066. [Google Scholar] [CrossRef]
- Haraguchi, N.; Kaseda, J.; Nakayama, Y.; Nagahama, K.; Ogawa, T.; Matsuoka, M. Characterization of mutants expressing thermostable D1 and D2 polypeptides of photosystem II in the cyanobacterium Synechococcus elongatus PCC 7942. J. Biosci. Bioeng. 2018, 126, 417–424. [Google Scholar] [CrossRef]
- Andrizhiyevskaya, E.G.; Frolov, D.; Van Grondelle, R.; Dekker, J.P. Energy transfer and trapping in the photosystem I complex of Synechococcus PCC 7942 and in its supercomplex with IsiA. Biochim. Biophys. Acta (BBA) Bioenerg. 2004, 1656, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.E.; Golden, S.S. Circadian rhythms in cyanobacteria. Microbiol. Mol. Biol. Rev. 2015, 79, 373–385. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Zhao, H.; Li, Z.; Tan, X.; Lu, X. Photosynthetic production of ethanol from carbon dioxide in genetically engineered cyanobacteria. Energy Environ. Sci. 2012, 5, 9857–9865. [Google Scholar] [CrossRef]
- Gao, X.; Gao, F.; Liu, D.; Zhang, H.; Nie, X.; Yang, C. Engineering the methylerythritol phosphate pathway in cyanobacteria for photosynthetic isoprene production from CO2. Energy Environ. Sci. 2016, 9, 1400–1411. [Google Scholar] [CrossRef]
- Sarnaik, A.; Nambissan, V.; Pandit, R.; Lali, A. Recombinant Synechococcus elongatus PCC 7942 for improved zeaxanthin production under natural light conditions. Algal Res. 2018, 36, 139–151. [Google Scholar] [CrossRef]
- Kusakabe, T.; Tatsuke, T.; Tsuruno, K.; Hirokawa, Y.; Atsumi, S.; Liao, J.C.; Hanai, T. Engineering a synthetic pathway in cyanobacteria for isopropanol production directly from carbon dioxide and light. Metab. Eng. 2013, 20, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Rubin, B.E.; Wetmore, K.M.; Price, M.N.; Diamond, S.; Shultzaberger, R.K.; Lowe, L.C.; Curtin, G.; Arkin, A.P.; Deutschbauer, A.; Golden, S.S. The essential gene set of a photosynthetic organism. Proc. Natl. Acad. Sci. USA 2015, 112, E6634–E6643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castenholz, R.W. Culturing methods for cyanobacteria. Methods Enzymol. 1988, 167, 68–93. [Google Scholar]
- Elhai, J.; Wolk, C.P. Conjugal transfer of DNA to cyanobacteria. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1988; Volume 167, pp. 747–754. [Google Scholar]
- Okada, N.; Ogawa, J.; Shima, J. Comprehensive analysis of genes involved in the oxidative stress tolerance using yeast heterozygous deletion collection. FEMS Yeast Res. 2014, 14, 425–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurokawa, M.; Seno, S.; Matsuda, H.; Ying, B.-W. Correlation between genome reduction and bacterial growth. DNA Res. 2016, 23, 517–525. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, M.; Ichimura, T.; Mizoguchi, H.; Tanaka, K.; Fujimitsu, K.; Keyamura, K.; Ote, T.; Yamakawa, T.; Yamazaki, Y.; Mori, H. Cell size and nucleoid organization of engineered Escherichia coli cells with a reduced genome. Mol. Microbiol. 2005, 55, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Ara, K.; Ozaki, K.; Nakamura, K.; Yamane, K.; Sekiguchi, J.; Ogasawara, N. Bacillus minimum genome factory: Effective utilization of microbial genome information. Biotechnol. Appl. Biochem. 2007, 46, 169–178. [Google Scholar]
- Reuß, D.R.; Altenbuchner, J.; Mäder, U.; Rath, H.; Ischebeck, T.; Sappa, P.K.; Thürmer, A.; Guérin, C.; Nicolas, P.; Steil, L.; et al. Large-scale reduction of the Bacillus subtilis genome: Consequences for the transcriptional network, resource allocation, and metabolism. Genome Res. 2017, 27, 289–299. [Google Scholar] [CrossRef] [Green Version]
- Wannier, T.M.; Kunjapur, A.M.; Rice, D.P.; McDonald, M.J.; Desai, M.M.; Church, G.M. Adaptive evolution of genomically recoded Escherichia coli. Proc. Natl. Acad. Sci. USA 2018, 115, 3090–3095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaCroix, R.A.; Sandberg, T.E.; O’Brien, E.J.; Utrilla, J.; Ebrahim, A.; Guzman, G.I.; Szubin, R.; Palsson, B.O.; Feist, A.M. Use of adaptive laboratory evolution to discover key mutations enabling rapid growth of Escherichia coli K-12 MG1655 on glucose minimal medium. Appl. Environ. Microbiol. 2015, 81, 17–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, F.; Ke, Z.; Xu, Y.; Wang, Y.; Zhu, G.; Gao, H.; Ji, S.; Xu, X. Systematic Large Fragment Deletions in the Genome of Synechococcus elongatus and the Consequent Changes in Transcriptomic Profiles. Genes 2023, 14, 1091. https://doi.org/10.3390/genes14051091
Hou F, Ke Z, Xu Y, Wang Y, Zhu G, Gao H, Ji S, Xu X. Systematic Large Fragment Deletions in the Genome of Synechococcus elongatus and the Consequent Changes in Transcriptomic Profiles. Genes. 2023; 14(5):1091. https://doi.org/10.3390/genes14051091
Chicago/Turabian StyleHou, Feifei, Zhufang Ke, Yi Xu, Yali Wang, Geqian Zhu, Hong Gao, Shuiling Ji, and Xudong Xu. 2023. "Systematic Large Fragment Deletions in the Genome of Synechococcus elongatus and the Consequent Changes in Transcriptomic Profiles" Genes 14, no. 5: 1091. https://doi.org/10.3390/genes14051091