

Embryonic Stem Cell-Derived Neurons as a Model System for Epigenome Maturation during Development
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Mouse Tissue Collection and In Vitro Neuron Differentiation
2.3. Murine Embryonic Stem Cell Culture and In Vitro Neuron Differentiation
2.4. Human In Vitro Neuron Differentiation
2.5. RT-qPCR
2.6. Immunocytochemistry
2.7. Immunohistochemistry
2.8. Optical Microscopy
2.9. Transmission Electron Microscopy
2.10. Whole Genome Bisulfite Sequencing
2.11. DNA Methylation Analysis
2.12. RNA-seq
2.13. Fluorescence-Activated Nuclear Sorting
2.14. Statistical Analysis
3. Results
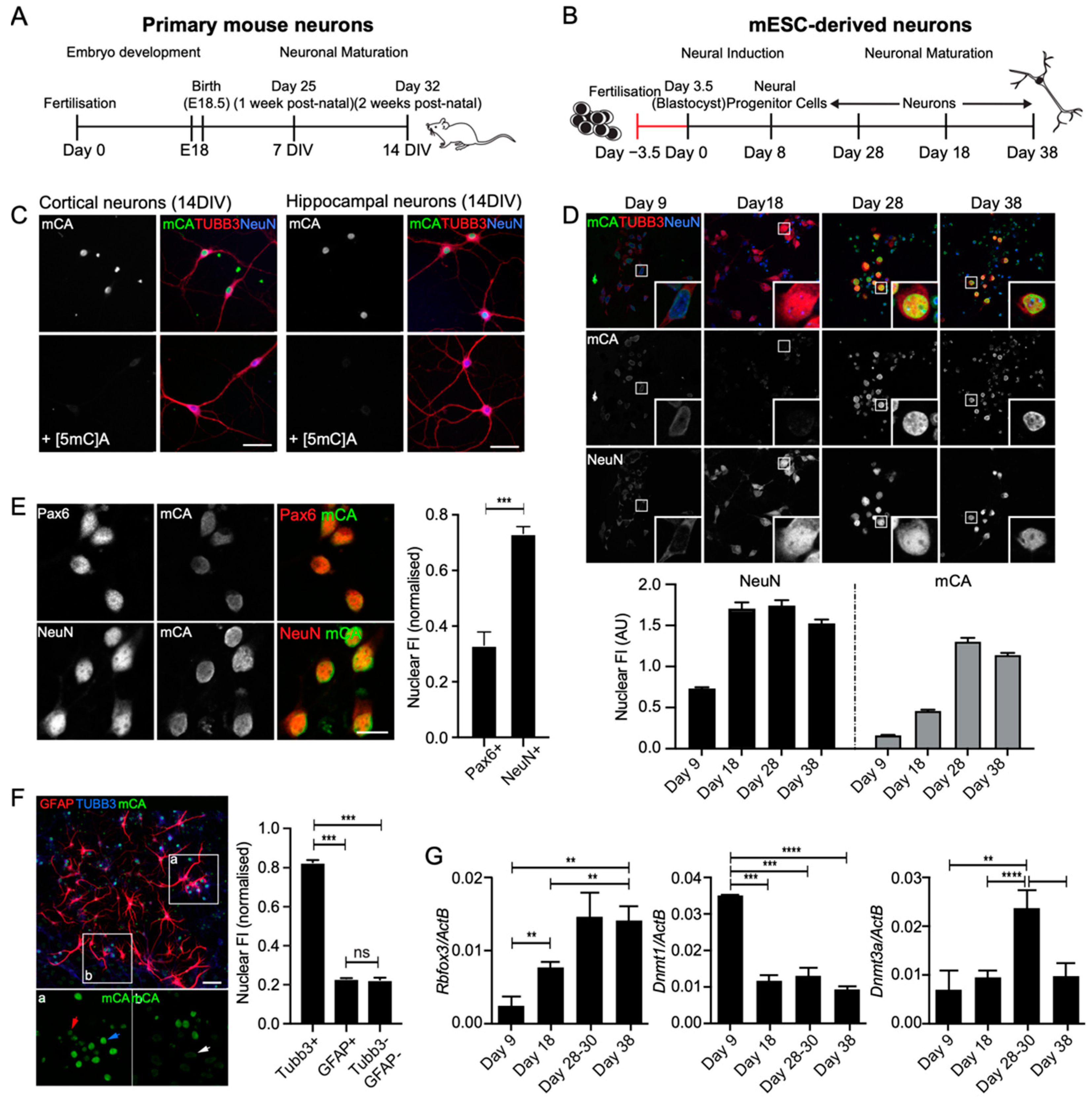
3.1. Immunocytochemical Labelling for mCA Accumulated in Postmitotic Neurons and Temporally Correlated with DNMT3a Expression
3.2. mCA Labelling Was Enriched at the Nuclear Periphery in Neurons
3.3. mESC-Derived Neuronal Cultures Acquired mCH to Levels Similar to Those Observed In Vivo
3.4. Global mCH and mCG Levels in mESC-Derived Neurons Revealed Hypermethylation Relative to In Vivo Adult Neurons
3.5. Genome-Wide Distribution of mCH and mCG between In Vivo and mESC-Derived Neurons Indicated Regional Hypermethylation
3.6. Similarities in Gene Regulation between In Vivo and In Vitro Neurons
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cortés-Mendoza, J.; Díaz de León-Guerrero, S.; Pedraza-Alva, G.; Pérez-Martínez, L. Shaping synaptic plasticity: The role of activity-mediated epigenetic regulation on gene transcription. Int. J. Dev. Neurosci. 2013, 31, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Day, J.J.; Childs, D.; Guzman-Karlsson, M.C.; Kibe, M.; Moulden, J.; Song, E.; Tahir, A.; Sweatt, J.D. DNA methylation regulates associative reward learning. Nat. Neurosci. 2013, 16, 1445–1452. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Zhou, Y.; Campbell, S.L.; Le, T.; Li, E.; Sweatt, J.D.; Silva, A.J.; Fan, G. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 2010, 13, 423–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gräff, J.; Woldemichael, B.T.; Berchtold, D.; Dewarrat, G.; Mansuy, I.M. Dynamic histone marks in the hippocampus and cortex facilitate memory consolidation. Nat. Commun. 2012, 3, 991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, C.A.; Sweatt, J.D. Covalent modification of DNA regulates memory formation. Neuron 2007, 53, 857–869. [Google Scholar] [CrossRef] [Green Version]
- Stroud, H.; Su, S.C.; Hrvatin, S.; Greben, A.W.; Renthal, W.; Boxer, L.D.; Nagy, M.A.; Hochbaum, D.R.; Kinde, B.; Gabel, H.W.; et al. Early-Life Gene Expression in Neurons Modulates Lasting Epigenetic States. Cell 2017, 171, 1151–1164.e16. [Google Scholar] [CrossRef]
- Bayraktar, G.; Kreutz, M.R. Neuronal DNA Methyltransferases: Epigenetic Mediators between Synaptic Activity and Gene Expression? Neuroscientist 2018, 24, 171–185. [Google Scholar] [CrossRef] [Green Version]
- Bayraktar, G.; Kreutz, M.R. The Role of Activity-Dependent DNA Demethylation in the Adult Brain and in Neurological Disorders. Front. Mol. Neurosci. 2018, 11, 169. [Google Scholar] [CrossRef] [Green Version]
- Fasolino, M.; Zhou, Z. The Crucial Role of DNA Methylation and MeCP2 in Neuronal Function. Genes 2017, 8, 141. [Google Scholar] [CrossRef] [Green Version]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.U.; Su, Y.; Shin, J.H.; Shin, J.; Li, H.; Xie, B.; Zhong, C.; Hu, S.; Le, T.; Fan, G.; et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci. 2014, 17, 215–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Ecker, J.R. Non-CG Methylation in the Human Genome. Annu. Rev. Genom. Hum. Genet. 2015, 16, 55–77. [Google Scholar] [CrossRef] [Green Version]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, W.; Barr, C.L.; Kim, A.; Yue, F.; Lee, A.Y.; Eubanks, J.; Dempster, E.L.; Ren, B. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell 2012, 148, 816–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kriaucionis, S.; Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef] [Green Version]
- Mellén, M.; Ayata, P.; Dewell, S.; Kriaucionis, S.; Heintz, N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 2012, 151, 1417–1430. [Google Scholar] [CrossRef] [Green Version]
- Szulwach, K.E.; Li, X.; Li, Y.; Song, C.-X.; Wu, H.; Dai, Q.; Irier, H.; Upadhyay, A.K.; Gearing, M.; Levey, A.I.; et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 2011, 14, 1607–1616. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.; Keown, C.L.; Kurihara, L.; Zhou, J.; He, Y.; Li, J.; Castanon, R.; Lucero, J.; Nery, J.R.; Sandoval, J.P.; et al. Single-cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. Science 2017, 357, 600–604. [Google Scholar] [CrossRef] [Green Version]
- Mo, A.; Mukamel, E.A.; Davis, F.P.; Luo, C.; Henry, G.L.; Picard, S.; Urich, M.A.; Nery, J.R.; Sejnowski, T.J.; Lister, R.; et al. Epigenomic Signatures of Neuronal Diversity in the Mammalian Brain. Neuron 2015, 86, 1369–1384. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Fan, G. The role of DNA methylation in the central nervous system and neuropsychiatric disorders. Int. Rev. Neurobiol. 2009, 89, 67–84. [Google Scholar]
- Feng, J.; Chang, H.; Li, E.; Fan, G. Dynamic expression of de novo DNA methyltransferases Dnmt3a and Dnmt3b in the central nervous system. J. Neurosci. Res. 2005, 79, 734–746. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, S.; Meletis, K.; Fu, D.; Jhaveri, S.; Jaenisch, R. Ablation of de novo DNA methyltransferase Dnmt3a in the nervous system leads to neuromuscular defects and shortened lifespan. Dev. Dyn. 2007, 236, 1663–1676. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, T.; Singh, A.K.; Chen, T. Genetic alterations of DNA methylation machinery in human diseases. Epigenomics 2015, 7, 247–265. [Google Scholar] [CrossRef] [PubMed]
- Ip, J.P.K.; Mellios, N.; Sur, M. Rett syndrome: Insights into genetic, molecular and circuit mechanisms. Nat. Rev. Neurosci. 2018, 19, 368–382. [Google Scholar] [CrossRef]
- Fath, T.; Ke, Y.D.; Hunning, P.; Götz, J.; Ittner, L.M. Primary support cultures of hippocampal and substantia nigra neurons. Nat. Protoc. 2009, 4, 78–85. [Google Scholar] [CrossRef]
- Nagy, A.; Rossant, J.; Nagy, R.; Abramow-Newerly, W.; Roder, J.C. Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc. Natl. Acad. Sci. USA 1993, 90, 8424–8428. [Google Scholar] [CrossRef] [Green Version]
- George, S.H.L.; Gertsenstein, M.; Vintersten, K.; Korets-Smith, E.; Murphy, J.; Stevens, M.E.; Haigh, J.J.; Nagy, A. Developmental and adult phenotyping directly from mutant embryonic stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 4455–4460. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.; Papadopulos, A.; Tomatis, V.M.; Sierecki, E.; Malintan, N.T.; Gormal, R.S.; Giles, N.; Johnston, W.A.; Alexandrov, K.; Gambin, Y.; et al. Increased polyubiquitination and proteasomal degradation of a Munc18-1 disease-linked mutant causes temperature-sensitive defect in exocytosis. Cell Rep. 2014, 9, 206–218. [Google Scholar] [CrossRef] [Green Version]
- Reinhardt, P.; Glatza, M.; Hemmer, K.; Tsytsyura, Y.; Thiel, C.S.; Höing, S.; Moritz, S.; Parga, J.A.; Wagner, L.; Bruder, J.M.; et al. Derivation and expansion using only small molecules of human neural progenitors for neurodegenerative disease modeling. PLoS ONE 2013, 8, e59252. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 2014, 9, 2329–2340. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Röhrl, C.; Meisslitzer-Ruppitsch, C.; Bittman, R.; Li, Z.; Pabst, G.; Prassl, R.; Strobl, J.; Neumüller, J.; Ellinger, A.; Pavelka, M.; et al. Combined Light and Electron Microscopy using Diaminobenzidine Photooxidation to Monitor Trafficking of Lipids Derived from Lipoprotein Particles. Curr. Pharm. Biotechnol. 2012, 13, 331–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, A.E.; Jones, T.R.; Lamprecht, M.R.; Clarke, C.; Kang, I.H.; Friman, O.; Guertin, D.A.; Chang, J.H.; Lindquist, R.A.; Moffat, J.; et al. CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006, 7, R100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.-M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinform. 2014, 47, 11.12.1–11.12.34. [Google Scholar] [CrossRef] [Green Version]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Michael Cherry, J.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- The Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, A.; Karpievitch, Y.V.; Eichten, S.R.; Borevitz, J.O.; Lister, R. HOME: A histogram based machine learning approach for effective identification of differentially methylated regions. BMC Bioinform. 2019, 20, 253. [Google Scholar] [CrossRef] [Green Version]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Baker-Andresen, D.; Zhao, Q.; Marshall, V.; Bredy, T.W. Methyl CpG binding domain ultra-sequencing: A novel method for identifying inter-individual and cell-type-specific variation in DNA methylation. Genes Brain Behav. 2014, 13, 721–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, S.; Saiwai, H.; Kumamaru, H.; Kubota, K.; Harada, A.; Yamaguchi, M.; Iwamoto, Y.; Ohkawa, Y. Flow cytometric sorting of neuronal and glial nuclei from central nervous system tissue. J. Cell. Physiol. 2011, 226, 552–558. [Google Scholar] [CrossRef]
- Deal, R.B.; Henikoff, S. The INTACT method for cell type-specific gene expression and chromatin profiling in Arabidopsis thaliana. Nat. Protoc. 2011, 6, 56–68. [Google Scholar] [CrossRef]
- Bibel, M.; Richter, J.; Lacroix, E.; Barde, Y.-A. Generation of a defined and uniform population of CNS progenitors and neurons from mouse embryonic stem cells. Nat. Protoc. 2007, 2, 1034–1043. [Google Scholar] [CrossRef]
- Cousin, M.A. Activity-dependent bulk synaptic vesicle endocytosis—A fast, high capacity membrane retrieval mechanism. Mol. Neurobiol. 2009, 39, 185–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullen, R.J.; Buck, C.R.; Smith, A.M. NeuN, a neuronal specific nuclear protein in vertebrates. Development 1992, 116, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Bibel, M.; Richter, J.; Schrenk, K.; Tucker, K.L.; Staiger, V.; Korte, M.; Goetz, M.; Barde, Y.-A. Differentiation of mouse embryonic stem cells into a defined neuronal lineage. Nat. Neurosci. 2004, 7, 1003–1009. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Damayanti, N.P.; Irudayaraj, J.; Dunn, K.; Zhou, F.C. Diversity of two forms of DNA methylation in the brain. Front. Genet. 2014, 5, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, C.; Lee, Q.Y.; Wapinski, O.; Castanon, R.; Nery, J.R.; Mall, M.; Kareta, M.S.; Cullen, S.M.; Goodell, M.A.; Chang, H.Y.; et al. Global DNA methylation remodeling during direct reprogramming of fibroblasts to neurons. eLife 2019, 8, e40197. [Google Scholar] [CrossRef] [PubMed]
- Arand, J.; Spieler, D.; Karius, T.; Branco, M.R.; Meilinger, D.; Meissner, A.; Jenuwein, T.; Xu, G.; Leonhardt, H.; Wolf, V.; et al. In vivo control of CpG and non-CpG DNA methylation by DNA methyltransferases. PLoS Genet. 2012, 8, e1002750. [Google Scholar] [CrossRef] [Green Version]
- Ramsahoye, B.H.; Biniszkiewicz, D.; Lyko, F.; Clark, V.; Bird, A.P.; Jaenisch, R. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc. Natl. Acad. Sci. USA 2000, 97, 5237–5242. [Google Scholar] [CrossRef] [Green Version]
- Liao, J.; Karnik, R.; Gu, H.; Ziller, M.J.; Clement, K.; Tsankov, A.M.; Akopian, V.; Gifford, C.A.; Donaghey, J.; Galonska, C.; et al. Targeted disruption of DNMT1, DNMT3A and DNMT3B in human embryonic stem cells. Nat. Genet. 2015, 47, 469–478. [Google Scholar] [CrossRef] [Green Version]
- Ziller, M.J.; Müller, F.; Liao, J.; Zhang, Y.; Gu, H.; Bock, C.; Boyle, P.; Epstein, C.B.; Bernstein, B.E.; Lengauer, T.; et al. Genomic distribution and inter-sample variation of non-CpG methylation across human cell types. PLoS Genet. 2011, 7, e1002389. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Yue, F.; McCleary, D.F.; Ye, Z.; Edsall, L.; Kuan, S.; Wagner, U.; Dixon, J.; Lee, L.; Lobanenkov, V.V.; et al. A map of the cis-regulatory sequences in the mouse genome. Nature 2012, 488, 116–120. [Google Scholar] [CrossRef] [Green Version]
- Hubbard, K.S.; Gut, I.M.; Lyman, M.E.; McNutt, P.M. Longitudinal RNA sequencing of the deep transcriptome during neurogenesis of cortical glutamatergic neurons from murine ESCs. F1000Research 2013, 2, 35. [Google Scholar] [CrossRef] [PubMed]
- Weyn-Vanhentenryck, S.M.; Feng, H.; Ustianenko, D.; Duffié, R.; Yan, Q.; Jacko, M.; Martinez, J.C.; Goodwin, M.; Zhang, X.; Hengst, U.; et al. Precise temporal regulation of alternative splicing during neural development. Nat. Commun. 2018, 9, 2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lienert, F.; Mohn, F.; Tiwari, V.K.; Baubec, T.; Roloff, T.C.; Gaidatzis, D.; Stadler, M.B.; Schübeler, D. Genomic prevalence of heterochromatic H3K9me2 and transcription do not discriminate pluripotent from terminally differentiated cells. PLoS Genet. 2011, 7, e1002090. [Google Scholar] [CrossRef]
- Tippmann, S.C.; Ivanek, R.; Gaidatzis, D.; Schöler, A.; Hoerner, L.; van Nimwegen, E.; Stadler, P.F.; Stadler, M.B.; Schübeler, D. Chromatin measurements reveal contributions of synthesis and decay to steady-state mRNA levels. Mol. Syst. Biol. 2012, 8, 593. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Pinto-Duarte, A.; Zander, M.; Lai, C.-Y.; Osteen, J.; Fang, L.; Luo, C.; Lucero, J.D.; Gomez-Castanon, R.; Nery, J.R.; et al. Dnmt3a knockout in excitatory neurons impairs postnatal synapse maturation and is partly compensated by repressive histone modification H3K27me3. bioRxiv 2021. [Google Scholar] [CrossRef] [Green Version]
- Ladd, A.N. CUG-BP, Elav-like family (CELF)-mediated alternative splicing regulation in the brain during health and disease. Mol. Cell. Neurosci. 2013, 56, 456–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Chen, K.; Lavery, L.A.; Baker, S.A. MeCP2 binds to non-CG methylated DNA as neurons mature, influencing transcription and the timing of onset for Rett syndrome. Proc. Natl. Acad. Sci. USA 2015, 112, 5509–5514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinde, B.; Wu, D.Y.; Greenberg, M.E.; Gabel, H.W. DNA methylation in the gene body influences MeCP2-mediated gene repression. Proc. Natl. Acad. Sci. USA 2016, 113, 15114–15119. [Google Scholar] [CrossRef] [Green Version]
- Lagger, S.; Connelly, J.C.; Schweikert, G.; Webb, S.; Selfridge, J.; Ramsahoye, B.H.; Yu, M.; He, C.; Sanguinetti, G.; Sowers, L.C.; et al. MeCP2 recognizes cytosine methylated tri-nucleotide and di-nucleotide sequences to tune transcription in the mammalian brain. PLoS Genet. 2017, 13, e1006793. [Google Scholar] [CrossRef]
- Skene, P.J.; Illingworth, R.S.; Webb, S.; Kerr, A.R.W.; James, K.D.; Turner, D.J.; Andrews, R.; Bird, A.P. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol. Cell 2010, 37, 457–468. [Google Scholar] [CrossRef] [Green Version]
- Stiles, J.; Jernigan, T.L. The basics of brain development. Neuropsychol. Rev. 2010, 20, 327–348. [Google Scholar] [CrossRef] [Green Version]
- Benito-Kwiecinski, S.; Lancaster, M.A. Brain Organoids: Human Neurodevelopment in a Dish. Cold Spring Harb. Perspect. Biol. 2020, 12, a035709. [Google Scholar] [CrossRef] [Green Version]
- Cakir, B.; Xiang, Y.; Tanaka, Y.; Kural, M.H.; Parent, M.; Kang, Y.-J.; Chapeton, K.; Patterson, B.; Yuan, Y.; He, C.-S.; et al. Engineering of human brain organoids with a functional vascular-like system. Nat. Methods 2019, 16, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Mansour, A.A.; Gonçalves, J.T.; Bloyd, C.W.; Li, H.; Fernandes, S.; Quang, D.; Johnston, S.; Parylak, S.L.; Jin, X.; Gage, F.H. An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 2018, 36, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Barry, C.; Schmitz, M.T.; Jiang, P.; Schwartz, M.P.; Duffin, B.M.; Swanson, S.; Bacher, R.; Bolin, J.M.; Elwell, A.L.; McIntosh, B.E.; et al. Species-specific developmental timing is maintained by pluripotent stem cells ex utero. Dev. Biol. 2017, 423, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Marchetto, M.C.; Hrvoj-Mihic, B.; Kerman, B.E.; Yu, D.X.; Vadodaria, K.C.; Linker, S.B.; Narvaiza, I.; Santos, R.; Denli, A.M.; Mendes, A.P.; et al. Species-specific maturation profiles of human, chimpanzee and bonobo neural cells. eLife 2019, 8, e37527. [Google Scholar] [CrossRef]
- Matsuda, M.; Hayashi, H.; Garcia-Ojalvo, J.; Yoshioka-Kobayashi, K.; Kageyama, R.; Yamanaka, Y.; Ikeya, M.; Toguchida, J.; Alev, C.; Ebisuya, M. Species-specific segmentation clock periods are due to differential biochemical reaction speeds. Science 2020, 369, 1450–1455. [Google Scholar] [CrossRef]
- Rayon, T.; Stamataki, D.; Perez-Carrasco, R.; Garcia-Perez, L.; Barrington, C.; Melchionda, M.; Exelby, K.; Lazaro, J.; Tybulewicz, V.L.J.; Fisher, E.M.C.; et al. Species-specific pace of development is associated with differences in protein stability. Science 2020, 369, eaba7667. [Google Scholar] [CrossRef]
- Gordon, A.; Yoon, S.-J.; Tran, S.S.; Makinson, C.D.; Park, J.Y.; Andersen, J.; Valencia, A.M.; Horvath, S.; Xiao, X.; Huguenard, J.R.; et al. Long-term maturation of human cortical organoids matches key early postnatal transitions. Nat. Neurosci. 2021, 24, 331–342. [Google Scholar] [CrossRef]
- Kang, H.J.; Kawasawa, Y.I.; Cheng, F.; Zhu, Y.; Xu, X.; Li, M.; Sousa, A.M.M.; Pletikos, M.; Meyer, K.A.; Sedmak, G.; et al. Spatio-temporal transcriptome of the human brain. Nature 2011, 478, 483–489. [Google Scholar] [CrossRef] [Green Version]
- Furlanis, E.; Scheiffele, P. Regulation of Neuronal Differentiation, Function, and Plasticity by Alternative Splicing. Annu. Rev. Cell Dev. Biol. 2018, 34, 451–469. [Google Scholar] [CrossRef]
- Gallegos, D.A.; Chan, U.; Chen, L.-F.; West, A.E. Chromatin Regulation of Neuronal Maturation and Plasticity. Trends Neurosci. 2018, 41, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Gabel, H.W.; Kinde, B.; Stroud, H.; Gilbert, C.S.; Harmin, D.A.; Kastan, N.R.; Hemberg, M.; Ebert, D.H.; Greenberg, M.E. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 2015, 522, 89–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuleger, N.; Robson, M.I.; Schirmer, E.C. The nuclear envelope as a chromatin organizer. Nucleus 2011, 2, 339–349. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.R.E.; Azuara, V.; Perry, P.; Sauer, S.; Dvorkina, M.; Jørgensen, H.; Roix, J.; McQueen, P.; Misteli, T.; Merkenschlager, M.; et al. Neural induction promotes large-scale chromatin reorganisation of the Mash1 locus. J. Cell Sci. 2006, 119, 132–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peric-Hupkes, D.; Meuleman, W.; Pagie, L.; Bruggeman, S.W.M.; Solovei, I.; Brugman, W.; Gräf, S.; Flicek, P.; Kerkhoven, R.M.; van Lohuizen, M.; et al. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol. Cell 2010, 38, 603–613. [Google Scholar] [CrossRef]
- Walczak, A.; Szczepankiewicz, A.A.; Ruszczycki, B.; Magalska, A.; Zamlynska, K.; Dzwonek, J.; Wilczek, E.; Zybura-Broda, K.; Rylski, M.; Malinowska, M.; et al. Novel higher-order epigenetic regulation of the Bdnf gene upon seizures. J. Neurosci. 2013, 33, 2507–2511. [Google Scholar] [CrossRef] [Green Version]
- Chahrour, M.; Zoghbi, H.Y. The story of Rett syndrome: From clinic to neurobiology. Neuron 2007, 56, 422–437. [Google Scholar] [CrossRef] [Green Version]
- Young, J.I.; Hong, E.P.; Castle, J.C.; Crespo-Barreto, J.; Bowman, A.B.; Rose, M.F.; Kang, D.; Richman, R.; Johnson, J.M.; Berget, S.; et al. Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc. Natl. Acad. Sci. USA 2005, 102, 17551–17558. [Google Scholar] [CrossRef] [Green Version]
- Guarda, A.; Bolognese, F.; Bonapace, I.M.; Badaracco, G. Interaction between the inner nuclear membrane lamin B receptor and the heterochromatic methyl binding protein, MeCP2. Exp. Cell Res. 2009, 315, 1895–1903. [Google Scholar] [CrossRef]
- Kishi, N.; Macklis, J.D. MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol. Cell. Neurosci. 2004, 27, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Yazdani, M.; Deogracias, R.; Guy, J.; Poot, R.A.; Bird, A.; Barde, Y.-A. Disease modeling using embryonic stem cells: MeCP2 regulates nuclear size and RNA synthesis in neurons. Stem Cells 2012, 30, 2128–2139. [Google Scholar] [CrossRef]
- Tan, H.K.; Wu, C.-S.; Li, J.; Tan, Z.H.; Hoffman, J.R.; Fry, C.J.; Yang, H.; Di Ruscio, A.; Tenen, D.G. DNMT3B shapes the mCA landscape and regulates mCG for promoter bivalency in human embryonic stem cells. Nucleic Acids Res. 2019, 47, 7460–7475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maunakea, A.K.; Chepelev, I.; Cui, K.; Zhao, K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 2013, 23, 1256–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, A.; Yitzhaky, A.; Bossel Ben-Moshe, N.; Domany, E. An accessible database for mouse and human whole transcriptome qPCR primers. Bioinformatics 2013, 29, 1355–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikuni, T.; Uesaka, N.; Okuno, H.; Hirai, H.; Deisseroth, K.; Bito, H.; Kano, M. Arc/Arg3.1 is a postsynaptic mediator of activity-dependent synapse elimination in the developing cerebellum. Neuron 2013, 78, 1024–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, S.; Poppe, D.; Olova, N.; O’Leary, C.; Ivanova, E.; Pflueger, J.; Dechka, J.; Simmons, R.K.; Cooper, H.M.; Reik, W.; et al. Embryonic Stem Cell-Derived Neurons as a Model System for Epigenome Maturation during Development. Genes 2023, 14, 957. https://doi.org/10.3390/genes14050957
Martin S, Poppe D, Olova N, O’Leary C, Ivanova E, Pflueger J, Dechka J, Simmons RK, Cooper HM, Reik W, et al. Embryonic Stem Cell-Derived Neurons as a Model System for Epigenome Maturation during Development. Genes. 2023; 14(5):957. https://doi.org/10.3390/genes14050957
Chicago/Turabian StyleMartin, Sally, Daniel Poppe, Nelly Olova, Conor O’Leary, Elena Ivanova, Jahnvi Pflueger, Jennifer Dechka, Rebecca K. Simmons, Helen M. Cooper, Wolf Reik, and et al. 2023. "Embryonic Stem Cell-Derived Neurons as a Model System for Epigenome Maturation during Development" Genes 14, no. 5: 957. https://doi.org/10.3390/genes14050957