
Complete Mitogenome and Phylogenetic Analysis of the Carthamus tinctorius L.


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials, Total DNA Extraction, and Genome Sequencing
2.2. Assembly and Annotation of Mitogenome
2.3. Repeats Identification and SSRs Analysis
2.4. Collinear Analysis and Rearrangement Analysis of Mitogenomes between Safflower and Two Other Asteraceae Species
2.5. Analysis of Codon Usage Bias Patterns for Protein-Coding Genes (PCGs)
2.6. Identification of RNA Editing Sites
2.7. Intracellular Gene Transfer Analysis
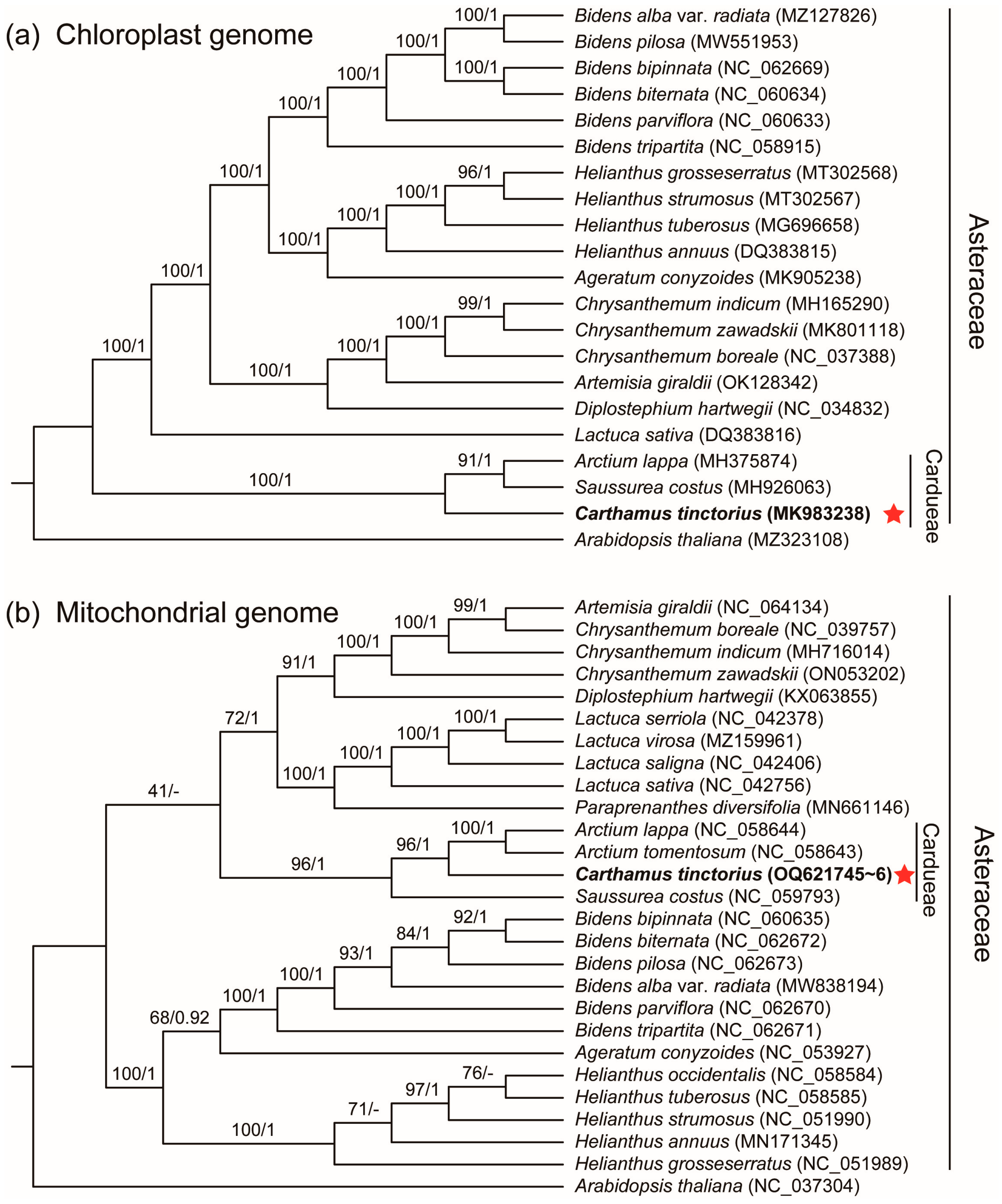
2.8. Phylogenetic Analysis
3. Results and Discussion
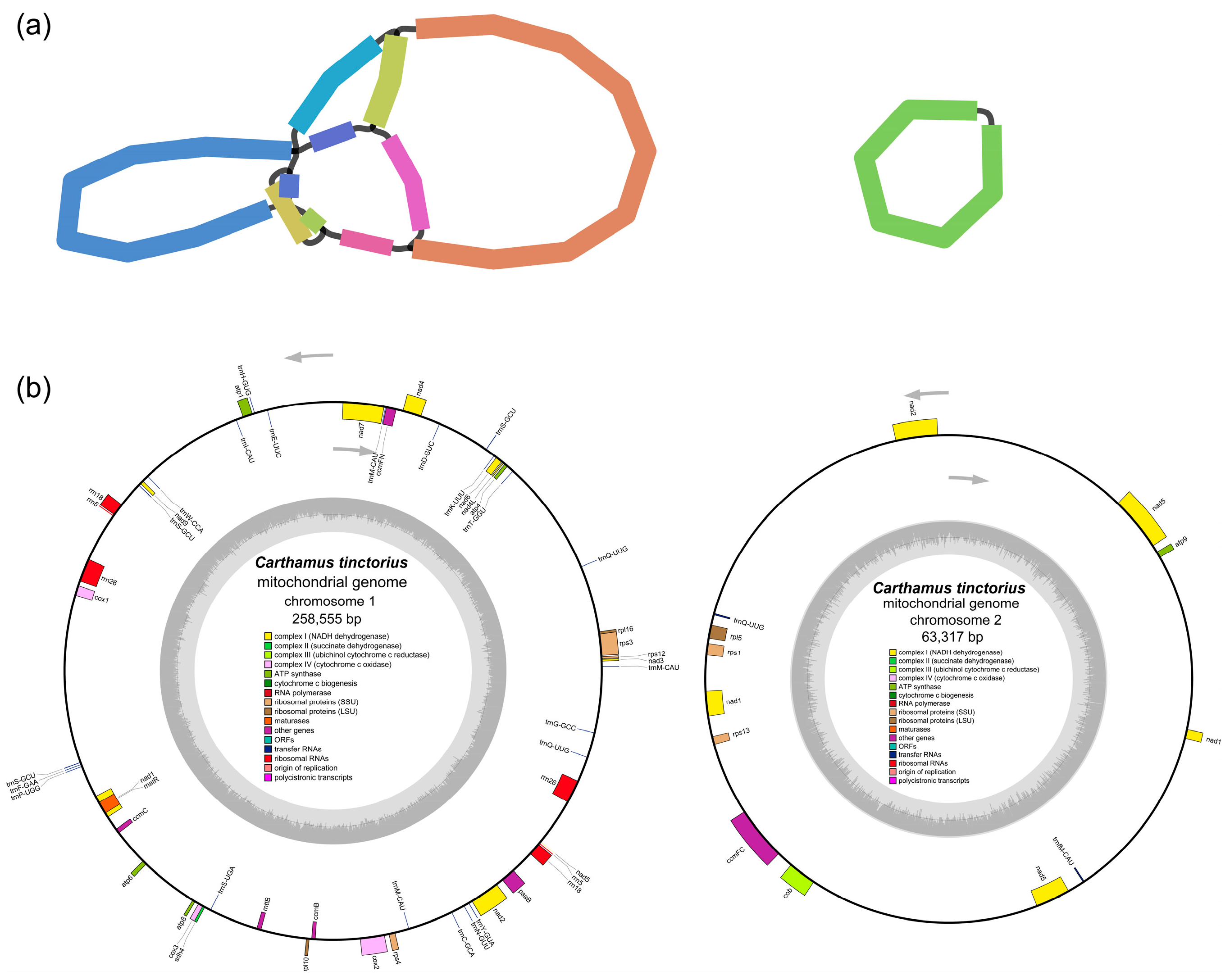
3.1. Mitogenome Characterization of C. tinctorius
3.2. Repeats, SSRs, and Collinear Analysis in the C. tinctorius Mitogenome
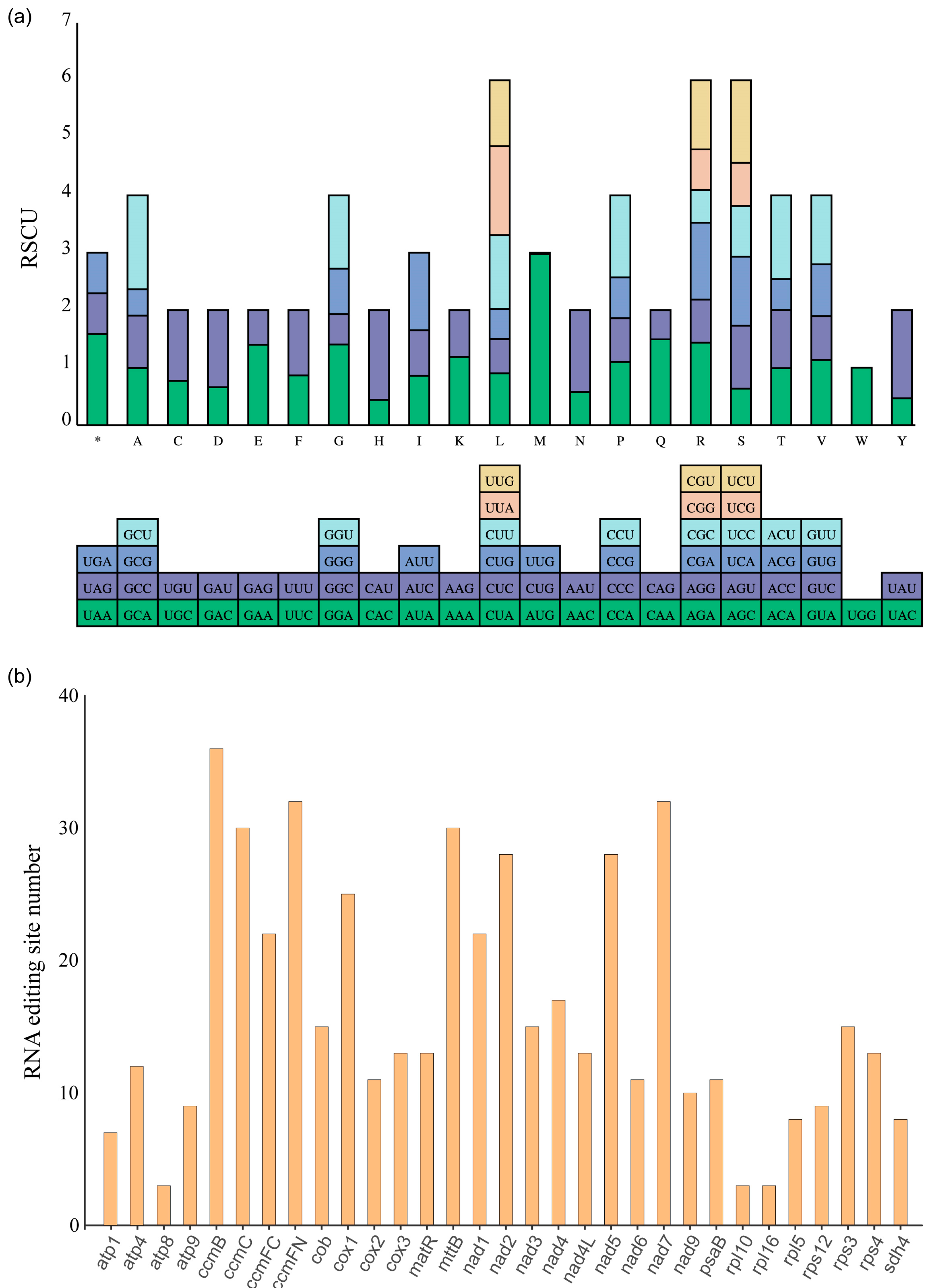
3.3. Codon Usage Bias Patterns and RNA Editing Sites Analysis for Protein-Coding Genes (PCGs)
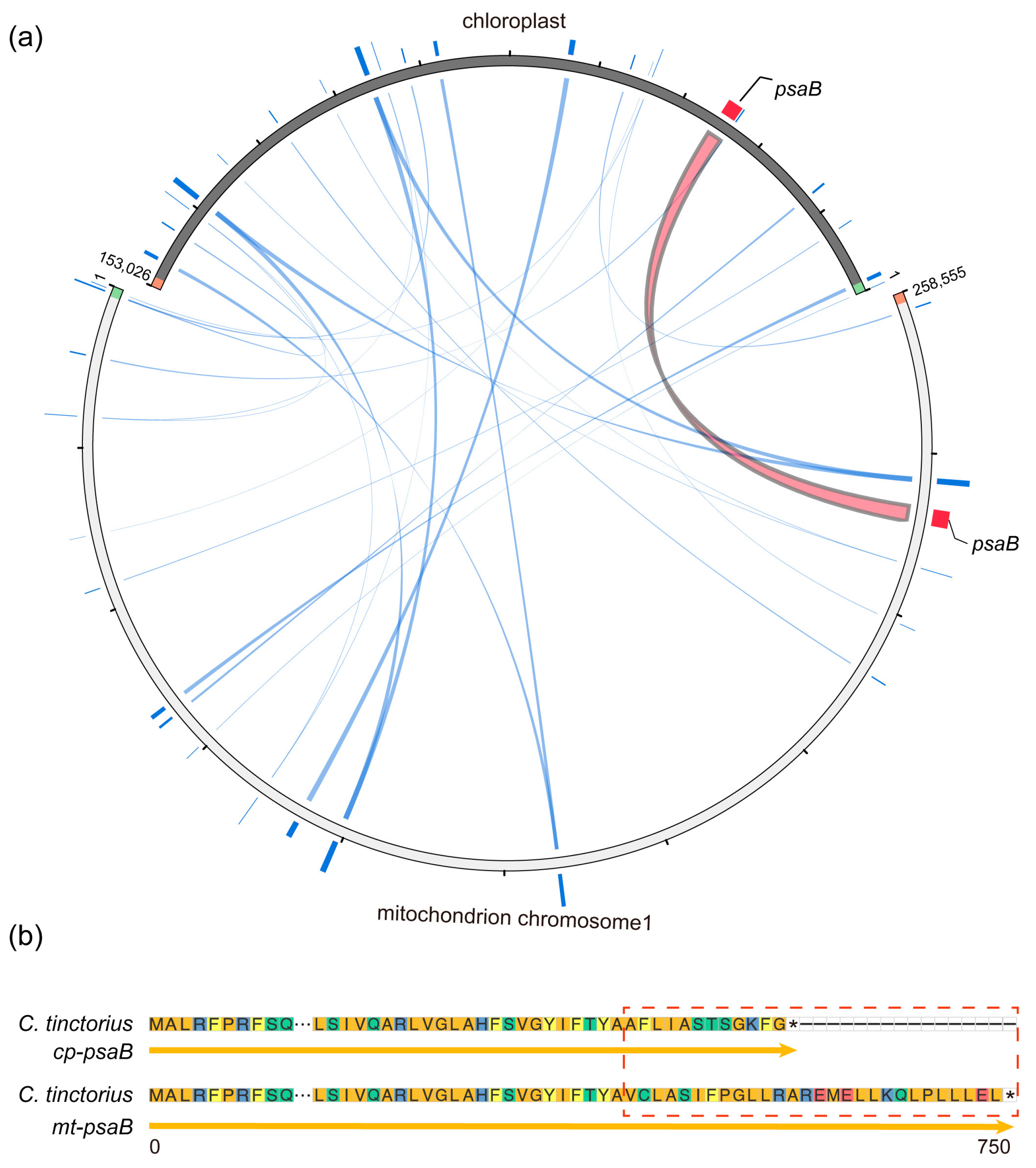
3.4. Intracellular Gene Transfer of C. tinctorius Organelle Genomes
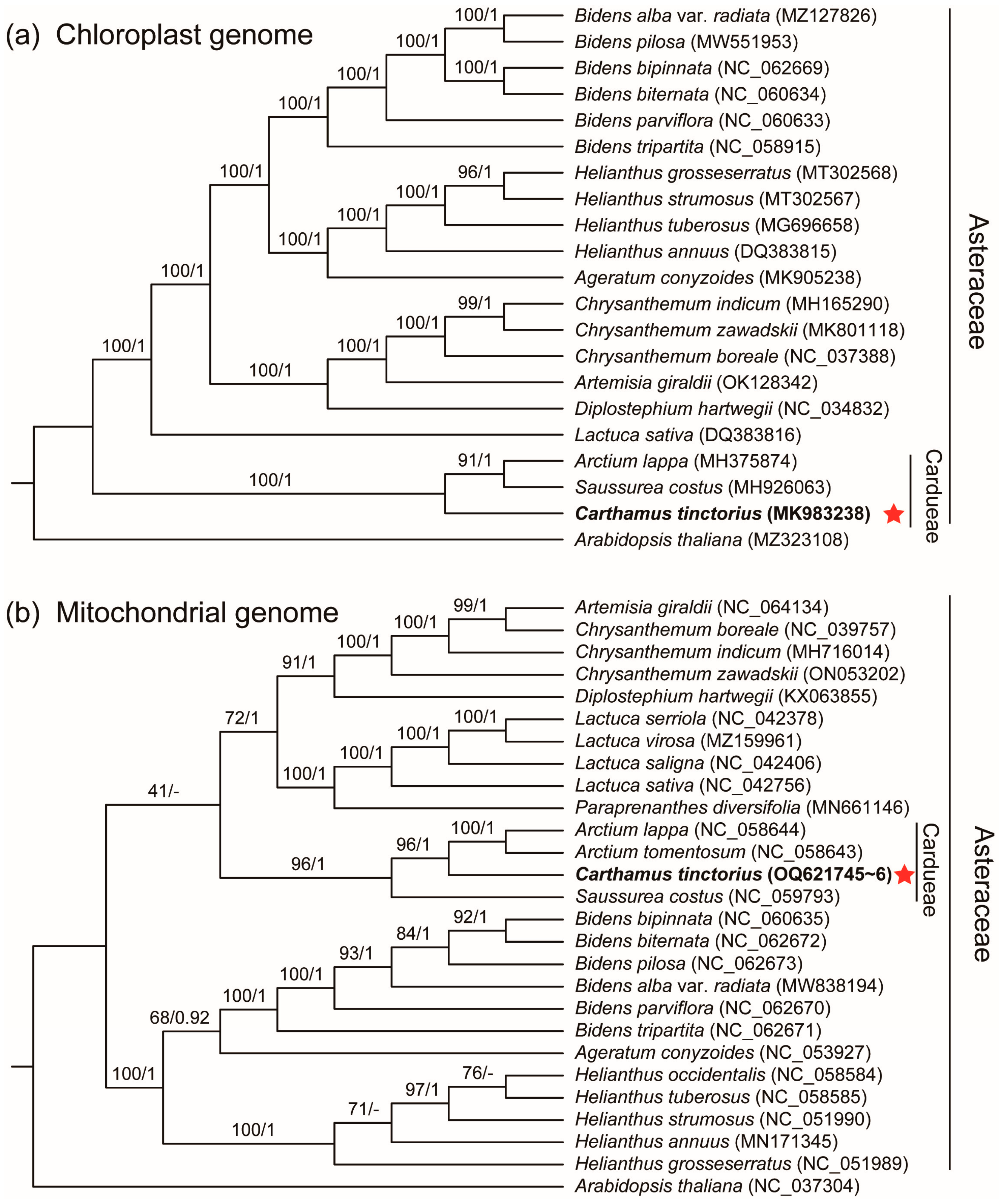
3.5. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kozik, A.; Rowan, B.A.; Lavelle, D.; Berke, L.; Schranz, M.E.; Michelmore, R.W.; Christensen, A.C. The alternative reality of plant mitochondrial DNA: One ring does not rule them all. PLoS Genet. 2019, 15, e1008373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyall, S.D.; Brown, M.T.; Johnson, P.J. Ancient invasions: From endosymbionts to organelles. Science 2004, 304, 253–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Grewe, F.; Fan, W.; Young, G.J.; Knoop, V.; Palmer, J.D.; Mower, J.P. Ginkgo and Welwitschia Mitogenomes Reveal Extreme Contrasts in Gymnosperm Mitochondrial Evolution. Mol. Biol. Evol. 2016, 33, 1448–1460. [Google Scholar] [CrossRef] [Green Version]
- Bullerwell, C.E.; Gray, M.W. Evolution of the mitochondrial genome: Protist connections to animals, fungi and plants. Curr. Opin. Microbiol. 2004, 7, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Knoop, V.; Volkmar, U.; Hecht, J.; Grewe, F. Mitochondrial Genome Evolution in the Plant Lineage. Plant Mitochondria 2011, 1, 3–29. [Google Scholar] [CrossRef]
- Smith, D.R. Genome evolution: Minicircular mtDNA and unusual heteroplasmy in a parasitic plant. Curr. Biol. 2022, 32, R86–R89. [Google Scholar] [CrossRef]
- Drouin, G.; Daoud, H.; Xia, J. Relative rates of synonymous substitutions in the mitochondrial, chloroplast and nuclear genomes of seed plants. Mol. Phylogenet. Evol. 2008, 49, 827–831. [Google Scholar] [CrossRef]
- Rodríguez-Moreno, L.; González, V.M.; Benjak, A.; Martí, M.C.; Puigdomènech, P.; Aranda, M.A.; Garcia-Mas, J. Determination of the melon chloroplast and mitochondrial genome sequences reveals that the largest reported mitochondrial genome in plants contains a significant amount of DNA having a nuclear origin. BMC Genom. 2011, 12, 424. [Google Scholar] [CrossRef] [Green Version]
- Putintseva, Y.A.; Bondar, E.I.; Simonov, E.P.; Sharov, V.V.; Oreshkova, N.V.; Kuzmin, D.A.; Konstantinov, Y.M.; Shmakov, V.N.; Belkov, V.I.; Sadovsky, M.G.; et al. Siberian larch (Larix sibirica Ledeb.) mitochondrial genome assembled using both short and long nucleotide sequence reads is currently the largest known mitogenome. BMC Genom. 2020, 21, 654. [Google Scholar] [CrossRef]
- Ma, J.; Wang, S.; Zhu, X.; Sun, G.; Chang, G.; Li, L.; Hu, X.; Zhang, S.; Zhou, Y.; Song, C.; et al. Major episodes of horizontal gene transfer drove the evolution of land plants. Mol. Plant 2022, 15, 857–871. [Google Scholar] [CrossRef]
- Gualberto, J.M.; Newton, K.J. Plant Mitochondrial Genomes: Dynamics and Mechanisms of Mutation. Annu. Rev. Plant Biol. 2017, 68, 225–252. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Deng, Y.; Wang, J.; Zhou, M.; Liao, L.; Wang, C.; Peng, C.; Li, Y. Hydroxysafflor yellow A, a natural compound from Carthamus tinctorius L with good effect of alleviating atherosclerosis. Phytomedicine 2021, 91, 153694. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Ren, C.; Xue, X.; Lu, H.; Wang, K.; Wu, L. Safflomin A: A novel chemical marker for Carthamus tinctorius L. (Safflower) monofloral honey. Food Chem. 2022, 366, 130584. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Ambreen, H.; Variath, M.T.; Rao, A.R.; Agarwal, M.; Kumar, A.; Goel, S.; Jagannath, A. Utilization of Molecular, Phenotypic, and Geographical Diversity to Develop Compact Composite Core Collection in the Oilseed Crop, Safflower (Carthamus tinctorius L.) through Maximization Strategy. Front. Plant Sci. 2016, 7, 1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zafari, M.; Ebadi, A.; Jahanbakhsh, S.; Sedghi, M. Safflower (Carthamus tinctorius) Biochemical Properties, Yield, and Oil Content Affected by 24-Epibrassinosteroid and Genotype under Drought Stress. J. Agric. Food Chem. 2020, 68, 6040–6047. [Google Scholar] [CrossRef] [PubMed]
- Lou, Z.Q.; Liu, M.L. A pharmacognostical study of the Chinese drug, Honghua, FLOS Carthami tinctorius. Acta Pharm. Sin. 1956, 4, 233–239. [Google Scholar]
- Zhao, X.; He, Y.; Zhang, Y.; Wan, H.; Wan, H.; Yang, J. Inhibition of Oxidative Stress: An Important Molecular Mechanism of Chinese Herbal Medicine (Astragalus membranaceus, Carthamus tinctorius L., Radix Salvia Miltiorrhizae, etc.) in the Treatment of Ischemic Stroke by Regulating the Antioxidant System. Oxid. Med. Cell. Longev. 2022, 2022, 1425369. [Google Scholar] [CrossRef]
- Li, L.; Liu, J.; Li, X.; Guo, Y.; Fan, Y.; Shu, H.; Wu, G.; Peng, C.; Xiong, L. Sesquiterpenoids from the Florets of Carthamus tinctorius (Safflower) and Their Anti-Atherosclerotic Activity. Nutrients 2022, 14, 5348. [Google Scholar] [CrossRef]
- Zhang, L.L.; Tian, K.; Tang, Z.H.; Chen, X.J.; Bian, Z.X.; Wang, Y.T.; Lu, J.J. Phytochemistry and Pharmacology of Carthamus tinctorius L. Am. J. Chin. Med. 2016, 44, 197–226. [Google Scholar] [CrossRef]
- Wu, Z.H.; Liao, R.; Dong, X.; Qin, R.; Liu, H. Complete chloroplast genome sequence of Carthamus tinctorius L. from PacBio Sequel Platform. Mitochondrial DNA Part B 2019, 4, 2635–2636. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Liu, H.; Zhan, W.; Yu, Z.; Qin, E.; Liu, S.; Yang, T.; Xiang, N.; Kudrna, D.; Chen, Y.; et al. The chromosome-scale reference genome of safflower (Carthamus tinctorius) provides insights into linoleic acid and flavonoid biosynthesis. Plant Biotechnol. J. 2021, 19, 1725–1742. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J. DNA Protocols for Plants. In Molecular Techniques in Taxonomy; Hewitt, G.M., Johnston, A.W.B., Young, J.P.W., Eds.; Springer: Berlin/Heidelberg, Germany, 1991; pp. 283–293. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Yu, W.; Yang, J.; Song, Y.; Depamphilis, C.W.; Yi, T.; Li, D. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [Green Version]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Yang, J.; Jing, Y.; Xu, L.; Yu, K.; Fang, X. NGenomeSyn: An easy-to-use and flexible tool for publication-ready visualization of syntenic relationships across multiple genomes. Bioinformatics 2023, 39, btad121. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, Y.; Li, R.; Li, X.; Chen, L.; Tang, J.; Qu, Y.; Yang, L.; Xin, P.; Li, Y. Analysis on codon usage bias of chloroplast genome in Mangifera indica. J. Cent. South Univ. For. Technol. 2021, 9, 148–156, 165. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.; Wang, G. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef]
- Wang, S.; Song, Q.; Li, S.; Hu, Z.; Dong, G.; Song, C.; Huang, H.; Liu, Y. Assembly of a Complete Mitogenome of Chrysanthemum nankingense Using Oxford Nanopore Long Reads and the Diversity and Evolution of Asteraceae Mitogenomes. Genes 2018, 9, 547. [Google Scholar] [CrossRef] [Green Version]
- Yue, J.; Lu, Q.; Ni, Y.; Chen, P.; Liu, C. Comparative analysis of the plastid and mitochondrial genomes of Artemisia giraldii Pamp. Sci. Rep. 2022, 12, 13931. [Google Scholar] [CrossRef]
- Zhang, C.; Huang, C.; Liu, M.; Hu, Y.; Panero, J.L.; Luebert, F.; Gao, T.; Ma, H. Phylotranscriptomic insights into Asteraceae diversity, polyploidy, and morphological innovation. J. Integr. Plant Biol. 2021, 63, 1273–1293. [Google Scholar] [CrossRef]
- Bi, C.; Lu, N.; Xu, Y.; He, C.; Lu, Z. Characterization and Analysis of the Mitochondrial Genome of Common Bean (Phaseolus vulgaris) by Comparative Genomic Approaches. Int. J. Mol. Sci. 2020, 21, 3778. [Google Scholar] [CrossRef]
- Guo, W.; Zhu, A.; Fan, W.; Mower, J.P. Complete mitochondrial genomes from the ferns Ophioglossum californicum and Psilotum nudum are highly repetitive with the largest organellar introns. New Phytol. 2017, 213, 391–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gualberto, J.M.; Mileshina, D.; Wallet, C.; Niazi, A.K.; Weber-Lotfi, F.; Dietrich, A. The plant mitochondrial genome: Dynamics and maintenance. Biochimie 2014, 100, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Alverson, A.J.; Wei, X.; Rice, D.W.; Stern, D.B.; Barry, K.; Palmer, J.D. Insights into the Evolution of Mitochondrial Genome Size from Complete Sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Mol. Biol. Evol. 2010, 27, 1436–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grewe, F.; Viehoever, P.; Weisshaar, B.; Knoop, V. A trans-splicing group I intron and tRNA-hyperediting in the mitochondrial genome of the lycophyte Isoetes engelmannii. Nucleic Acids Res. 2009, 37, 5093–5104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Tang, H.; Luo, H.; Tang, J.; Zhong, N.; Xiao, L. Complete mitochondrial genome assembly and comparison of Camellia sinensis var. Assamica cv. Duntsa. Front. Plant Sci. 2023, 14, 1117002. [Google Scholar] [CrossRef]
- Yang, J.X.; Dierckxsens, N.; Bai, M.Z.; Guo, Y.Y. Multichromosomal Mitochondrial Genome of Paphiopedilum micranthum: Compact and Fragmented Genome, and Rampant Intracellular Gene Transfer. Int. J. Mol. Sci. 2023, 24, 3976. [Google Scholar] [CrossRef]
- Liao, X.; Zhao, Y.; Kong, X.; Khan, A.; Zhou, B.; Liu, D.; Kashif, M.H.; Chen, P.; Wang, H.; Zhou, R. Complete sequence of kenaf (Hibiscus cannabinus) mitochondrial genome and comparative analysis with the mitochondrial genomes of other plants. Sci. Rep. 2018, 8, 12714. [Google Scholar] [CrossRef] [Green Version]
- Straub, S.C.; Cronn, R.C.; Edwards, C.; Fishbein, M.; Liston, A. Horizontal transfer of DNA from the mitochondrial to the plastid genome and its subsequent evolution in milkweeds (apocynaceae). Genome Biol. Evol. 2013, 5, 1872–1885. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequences | Chromosome 1 | Chromosome 2 |
|---|---|---|
| Length (bp) | 258,555 | 63,317 |
| Gene name | atp1, atp4, atp6, atp8, ccmB, ccmC, ccmFN, cox1, cox2, cox3, matR, mttB, nad1, nad2, nad3, nad4, nad4L, nad4, nad5, nad6, nad7, nad9, psaB, rpl10, rpl16, rps12, rps3, rps4, rrn18, rrn26, rrn5, sdh4, trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnG-GCC, trnH-GUG, trnI-CAU, trnK-UUU, trnM-CAU, trnN-GUU, trnP-UGG, trnQ-UUG, trnS-GCU, trnS-UGA, trnT-GGU, trnW-CCA, trnY-GUA | atp9, ccmFC, cob, nad1, nad2, nad5, rpl5, rps1, rps13, trnQ-UUG, trnfM-CAU |
| Gene number | 48 | 11 |
| GC content | 45.24% | 45.43% |
| Functional Classification of Genes | Gene Name |
|---|---|
| ATP synthase | atp1, atp4, atp6, atp8, atp9 |
| Cytochrome c biogenesis | ccmB, ccmC, ccmFc *, ccmFn |
| Ubichinol cytochrome c reductase | cob |
| Cytochrome c oxidase | cox1, cox2 **, cox3 |
| Maturases | matR |
| Transport membrance protein | mttB |
| NADH dehydrogenase | nad1 **, nad2 ***, nad3, nad4 *, nad4L, nad5 **, nad6, nad7 ****, nad9 |
| Ribosomal proteins (LSU) | rpl10, rpl16, rpl5 |
| Ribosomal proteins (SSU) | rps12, rps3 *, rps4 |
| Succinate dehydrogenase | sdh4 |
| Ribosomal RNAs | rrn18 (×2), rrn26 (×2), rrn5 (×2) |
| Transfer RNAs | trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnG-GCC, trnH-GUG, trnK-UUU, trnM-CAT, trnM-CAU (×3), trnN-GUU, trnP-UGG, trnQ-UUG (×2), trnS-GCU (×3), trnS-UGA, trnT-GGU, trnT-UGU, trnW-CCA, trnY-GUA |
| Photosystem I | psaB |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Z.; Yang, T.; Qin, R.; Liu, H. Complete Mitogenome and Phylogenetic Analysis of the Carthamus tinctorius L. Genes 2023, 14, 979. https://doi.org/10.3390/genes14050979
Wu Z, Yang T, Qin R, Liu H. Complete Mitogenome and Phylogenetic Analysis of the Carthamus tinctorius L. Genes. 2023; 14(5):979. https://doi.org/10.3390/genes14050979
Chicago/Turabian StyleWu, Zhihua, Tiange Yang, Rui Qin, and Hong Liu. 2023. "Complete Mitogenome and Phylogenetic Analysis of the Carthamus tinctorius L." Genes 14, no. 5: 979. https://doi.org/10.3390/genes14050979
APA StyleWu, Z., Yang, T., Qin, R., & Liu, H. (2023). Complete Mitogenome and Phylogenetic Analysis of the Carthamus tinctorius L. Genes, 14(5), 979. https://doi.org/10.3390/genes14050979