
The Low-Copy-Number Satellite DNAs of the Model Beetle Tribolium castaneum
 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Insect Material and DNA Extraction
2.2. Illumina Sequencing and Graph-Based Clustering of Sequencing Reads
2.3. Computational Analyses of satDNAs
2.4. SatDNA Amplification, Cloning, and Sequencing
2.5. DNA Probes
2.6. Southern Blot Hybridization
2.7. Chromosome Preparations and Fluorescence In Situ Hybridization (FISH)
3. Results
3.1. High-Throughput Search for satDNAs
3.2. Characteristics of the Low-Copy satDNAs
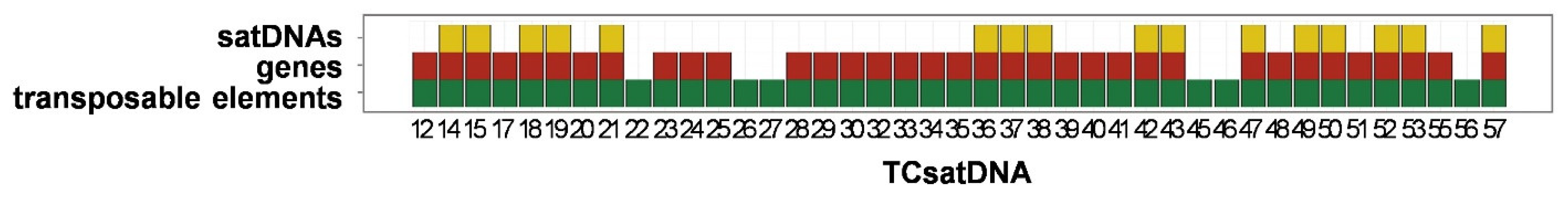
3.3. Organization of the Low-Copy satDNAs
3.4. Experimental Analysis of the Three Low-Copy satDNAs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kit, S. Equilibrium Sedimentation in Density Gradients of DNA Preparations from Animal Tissues. J. Mol. Biol. 1961, 3, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Talbert, P.B.; Henikoff, S. The Genetics and Epigenetics of Satellite Centromeres. Genome Res. 2022, 32, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Nergadze, S.G.; Piras, F.M.; Gamba, R.; Corbo, M.; Cerutti, F.; McCarter, J.G.W.; Cappelletti, E.; Gozzo, F.; Harman, R.M.; Antczak, D.F.; et al. Birth, Evolution, and Transmission of Satellite-Free Mammalian Centromeric Domains. Genome Res. 2018, 28, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Feliciello, I.; Akrap, I.; Ugarković, Đ. Satellite DNA Modulates Gene Expression in the Beetle Tribolium Castaneum after Heat Stress. PLoS Genet. 2015, 11, e1005466. [Google Scholar] [CrossRef]
- Nogalski, M.T.; Shenk, T. HSATII RNA Is Induced via a Noncanonical ATM-Regulated DNA Damage Response Pathway and Promotes Tumor Cell Proliferation and Movement. Proc. Natl. Acad. Sci. USA 2020, 117, 31891–31901. [Google Scholar] [CrossRef]
- Lopes, M.; Louzada, S.; Ferreira, D.; Veríssimo, G.; Eleutério, D.; Gama-Carvalho, M.; Chaves, R. Human Satellite 1A Analysis Provides Evidence of Pericentromeric Transcription. BMC Biol. 2023, 21, 28. [Google Scholar] [CrossRef]
- Garrido-Ramos, M.A. Satellite DNA: An Evolving Topic. Genes 2017, 8, 230. [Google Scholar] [CrossRef]
- Kuhn, G.C.S.; Küttler, H.; Moreira-Filho, O.; Heslop-Harrison, J.S. The 1.688 Repetitive DNA of Drosophila: Concerted Evolution at Different Genomic Scales and Association with Genes. Mol. Biol. Evol. 2012, 29, 7–11. [Google Scholar] [CrossRef]
- Pavlek, M.; Gelfand, Y.; Plohl, M.; Meštrović, N. Genome-Wide Analysis of Tandem Repeats in Tribolium Castaneum Genome Reveals Abundant and Highly Dynamic Tandem Repeat Families with Satellite DNA Features in Euchromatic Chromosomal Arms. DNA Res. 2015, 22, 387–401. [Google Scholar] [CrossRef]
- Novák, P.; Neumann, P.; Pech, J.; Steinhaisl, J.; MacAs, J. RepeatExplorer: A Galaxy-Based Web Server for Genome-Wide Characterization of Eukaryotic Repetitive Elements from Next-Generation Sequence Reads. Bioinformatics 2013, 29, 792–793. [Google Scholar] [CrossRef]
- Novák, P.; Neumann, P.; Macas, J. Global Analysis of Repetitive DNA from Unassembled Sequence Reads Using RepeatExplorer2. Nat. Protoc. 2020, 15, 3745–3776. [Google Scholar] [CrossRef]
- Novák, P.; Robledillo, L.Á.; Koblížková, A.; Vrbová, I.; Neumann, P.; Macas, J. TAREAN: A Computational Tool for Identification and Characterization of Satellite DNA from Unassembled Short Reads. Nucleic Acids Res. 2017, 45, e111. [Google Scholar] [CrossRef]
- Utsunomia, R.; Silva, D.M.Z.d.A.; Ruiz-Ruano, F.J.; Goes, C.A.G.; Melo, S.; Ramos, L.P.; Oliveira, C.; Porto-Foresti, F.; Foresti, F.; Hashimoto, D.T. Satellitome Landscape Analysis of Megaleporinus Macrocephalus (Teleostei, Anostomidae) Reveals Intense Accumulation of Satellite Sequences on the Heteromorphic Sex Chromosome. Sci. Rep. 2019, 9, 5856. [Google Scholar] [CrossRef]
- Boštjančić, L.L.; Bonassin, L.; Anušić, L.; Lovrenčić, L.; Besendorfer, V.; Maguire, I.; Grandjean, F.; Austin, C.M.; Greve, C.; Hamadou, A.B.; et al. The Pontastacus Leptodactylus (Astacidae) Repeatome Provides Insight into Genome Evolution and Reveals Remarkable Diversity of Satellite DNA. Front. Genet. 2021, 11, 611745. [Google Scholar] [CrossRef]
- Šatović-Vukšić, E.; Plohl, M. Satellite DNAs—From Localized to Highly Dispersed Genome Components. Genes 2023, 14, 742. [Google Scholar] [CrossRef]
- Ruiz-Ruano, F.J.; López-León, M.D.; Cabrero, J.; Camacho, J.P.M. High-Throughput Analysis of the Satellitome Illuminates Satellite DNA Evolution. Sci. Rep. 2016, 6, 28333. [Google Scholar] [CrossRef]
- Altemose, N.; Logsdon, G.A.; Bzikadze, A.V.; Sidhwani, P.; Langley, S.A.; Caldas, G.V.; Hoyt, S.J.; Uralsky, L.; Ryabov, F.D.; Shew, C.J.; et al. Complete Genomic and Epigenetic Maps of Human Centromeres. Science 2022, 376, 6588. [Google Scholar] [CrossRef]
- Dover, G.A. Molecular Drive in Multigene Families: How Biological Novelties Arise, Spread and Are Assimilated. Trends Genet. 1986, 2, 159–165. [Google Scholar] [CrossRef]
- Fry, K.; Salser, W. Nucleotide Sequences of HS-α Satellite DNA from Kangaroo Rat Dipodomys Ordii and Characterization of Similar Sequences in Other Rodents. Cell 1977, 12, 1069–1084. [Google Scholar] [CrossRef]
- Meštrović, N.; Plohl, M.; Mravinac, B.; Ugarković, Đ. Evolution of Satellite DNAs from the Genus Palorus—Experimental Evidence for the “Library” Hypothesis. Mol. Biol. Evol. 1998, 15, 1062–1068. [Google Scholar] [CrossRef]
- Camacho, J.P.M.; Cabrero, J.; López-León, M.D.; Martín-Peciña, M.; Perfectti, F.; Garrido-Ramos, M.A.; Ruiz-Ruano, F.J. Satellitome Comparison of Two Oedipodine Grasshoppers Highlights the Contingent Nature of Satellite DNA Evolution. BMC Biol. 2022, 20, 36. [Google Scholar] [CrossRef]
- Sokoloff, A. The Biology of Tribolium, with Special Emphasis on Genetic Aspects; Clarendon Press: Oxford, UK, 1972; Volume 1, ISBN 0198573537. [Google Scholar]
- Campbell, J.F.; Athanassiou, C.G.; Hagstrum, D.W.; Zhu, K.Y. Tribolium Castaneum: A Model Insect for Fundamental and Applied Research. Annu. Rev. Entomol. 2022, 67, 347–365. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Gibbs, R.A.; Weinstock, G.M.; Brown, S.; Denell, R.; Beeman, R.W.; Gibbs, R.; Bucher, G.; Friedrich, M.; Grimmelikhuijzen, C.J.P.; et al. The Genome of the Model Beetle and Pest Tribolium Castaneum. Nature 2008, 452, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Herndon, N.; Shelton, J.; Gerischer, L.; Ioannidis, P.; Ninova, M.; Dönitz, J.; Waterhouse, R.M.; Liang, C.; Damm, C.; Siemanowski, J.; et al. Enhanced Genome Assembly and a New Official Gene Set for Tribolium Castaneum. BMC Genom. 2020, 21, 47. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.J.; Henry, J.K.; Black IV, W.C.; Denell, R.E. Molecular Genetic Manipulation of the Red Flour Beetle: Genome Organization and Cloning of a Ribosomal Protein Gene. Insect Biochem. 1990, 20, 185–193. [Google Scholar] [CrossRef]
- Wang, S.; Lorenzen, M.D.; Beeman, R.W.; Brown, S.J. Analysis of Repetitive DNA Distribution Patterns in the Tribolium Castaneum Genome. Genome Biol. 2008, 9, R61. [Google Scholar] [CrossRef]
- Ugarković, D.; Podnar, M.; Plohl, M. Satellite DNA of the Red Flour Beetle Tribolium Castaneum—Comparative Study of Satellites from the Genus Tribolium. Mol. Biol. Evol. 1996, 13, 1059–1066. [Google Scholar] [CrossRef]
- Gržan, T.; Despot-Slade, E.; Meštrović, N.; Plohl, M.; Mravinac, B. CenH3 Distribution Reveals Extended Centromeres in the Model Beetle Tribolium Castaneum. PLoS Genet. 2020, 16, e1009115. [Google Scholar] [CrossRef]
- Feliciello, I.; Akrap, I.; Brajkovi, J.; Zlatar, I.; Ugarkovic, D. Satellite DNA as a Driver of Population Divergence in the Red Flour Beetle Tribolium Castaneum. Genome Biol. Evol. 2015, 7, 228–239. [Google Scholar] [CrossRef]
- Andrews, S.; Babraham, I. FastQC: A Quality Control Tool for High throughput Sequence Data. 2013. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 25 August 2022).
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a Database of Repetitive Elements in Eukaryotic Genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef]
- Kohany, O.; Gentles, A.J.; Hankus, L.; Jurka, J. Annotation, Submission and Screening of Repetitive Elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinform. 2006, 7, 474. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Muscle: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Benoit, M.; Drost, H.G. A Predictive Approach to Infer the Activity and Natural Variation of Retrotransposon Families in Plants. Methods Mol. Biol. 2021, 2250, 1–14. [Google Scholar] [PubMed]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. 2015. Available online: http://www.repeatmasker.org/ (accessed on 11 October 2022).
- Kõressaar, T.; Lepamets, M.; Kaplinski, L.; Raime, K.; Andreson, R.; Remm, M. Primer3-masker: Integrating Masking of Template Sequence with Primer Design Software. Bioinformatics 2018, 34, 1937–1938. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Juan, C.; Vazquez, P.; Rubio, J.M.; Petitpierre, E.; Hewitt, G.M. Presence of Highly Repetitive DNA Sequences in Tribolium Flour-Beetles. Heredity 1993, 70, 1–8. [Google Scholar] [CrossRef]
- Stuart, J.J.; Mocelin, G. Cytogenetics of Chromosome Rearrangements in Tribolium Castaneum. Genome 1995, 38, 673–680. [Google Scholar] [CrossRef]
- Plohl, M.; Lucijanic-Justic, V.; Ugarkovic, D.; Petitpierre, E.; Juan, C. Satellite DNA and Heterochromatin of the Flour Beetle Tribolium Confusum. Genome 1993, 36, 467–475. [Google Scholar] [CrossRef]
- Ugarković, D.; Durajlija, S.; Plohl, M. Evolution of Tribolium Madens (Insecta, Coleoptera) Satellite DNA through DNA Inversion and Insertion. J. Mol. Evol. 1996, 42, 350–358. [Google Scholar] [CrossRef]
- Mravinac, B.; Plohl, M.; Ugarković, D. Conserved Patterns in the Evolution of Tribolium Satellite DNAs. Gene 2004, 332, 169–177. [Google Scholar] [CrossRef]
- Mravinac, B.; Ugarković, D.; Franjević, D.; Plohl, M. Long Inversely Oriented Subunits Form a Complex Monomer of Tribolium Brevicornis Satellite DNA. J. Mol. Evol. 2005, 60, 513–525. [Google Scholar] [CrossRef]
- Mravinac, B.; Plohl, M. Parallelism in Evolution of Highly Repetitive DNAs in Sibling Species. Mol. Biol. Evol. 2010, 27, 1857–1867. [Google Scholar] [CrossRef]
- Mora, P.; Vela, J.; Ruiz-Ruano, F.J.; Ruiz-Mena, A.; Montiel, E.E.; Palomeque, T.; Lorite, P. Satellitome Analysis in the Ladybird Beetle Hippodamia Variegata (Coleoptera, Coccinellidae). Genes 2020, 11, 783. [Google Scholar] [CrossRef]
- Montiel, E.E.; Panzera, F.; Palomeque, T.; Lorite, P.; Pita, S. Satellitome Analysis of Rhodnius Prolixus, One of the Main Chagas Disease Vector Species. Int. J. Mol. Sci. 2021, 22, 6052. [Google Scholar] [CrossRef]
- Ruiz-Ruano, F.J.; Castillo-Martínez, J.; Cabrero, J.; Gómez, R.; Camacho, J.P.M.; López-León, M.D. High-Throughput Analysis of Satellite DNA in the Grasshopper Pyrgomorpha Conica Reveals Abundance of Homologous and Heterologous Higher-Order Repeats. Chromosoma 2018, 127, 323–340. [Google Scholar] [CrossRef]
- Montiel, E.E.; Mora, P.; Rico-Porras, J.M.; Palomeque, T.; Lorite, P. Satellitome of the Red Palm Weevil, Rhynchophorus Ferrugineus (Coleoptera: Curculionidae), the Most Diverse Among Insects. Front. Ecol. Evol. 2022, 10, 826808. [Google Scholar] [CrossRef]
- Mora, P.; Pita, S.; Montiel, E.E.; Rico-Porras, J.M.; Palomeque, T.; Panzera, F.; Lorite, P. Making the Genome Huge: The Case of Triatoma Delpontei, a Triatominae Species with More than 50% of Its Genome Full of Satellite DNA. Genes 2023, 14, 371. [Google Scholar] [CrossRef]
- Majid, M.; Yuan, H. Comparative Analysis of Transposable Elements in Genus Calliptamus Grasshoppers Revealed That Satellite Dna Contributes to Genome Size Variation. Insects 2021, 12, 837. [Google Scholar] [CrossRef]
- Talbert, P.B.; Henikoff, S. What Makes a Centromere? Exp. Cell Res. 2020, 389, 111895. [Google Scholar] [CrossRef]
- Heslop-Harrison, J.S.; Schwarzacher, T. Nucleosomes and Centromeric DNA Packaging. Proc. Natl. Acad. Sci. USA 2013, 110, 19974–19975. [Google Scholar] [CrossRef] [PubMed]
- Melters, D.P.; Bradnam, K.R.; Young, H.A.; Telis, N.; May, M.R.; Ruby, J.G.; Sebra, R.; Peluso, P.; Eid, J.; Rank, D.; et al. Comparative Analysis of Tandem Repeats from Hundreds of Species Reveals Unique Insights into Centromere Evolution. Genome Biol. 2013, 14, R10. [Google Scholar] [CrossRef] [PubMed]
- Ávila Robledillo, L.; Koblížková, A.; Novák, P.; Böttinger, K.; Vrbová, I.; Neumann, P.; Schubert, I.; Macas, J. Satellite DNA in Vicia Faba Is Characterized by Remarkable Diversity in Its Sequence Composition, Association with Centromeres, and Replication Timing. Sci. Rep. 2018, 8, 5838. [Google Scholar] [CrossRef] [PubMed]
- Vojvoda Zeljko, T.; Pavlek, M.; Meštrović, N.; Plohl, M. Satellite DNA-like Repeats Are Dispersed throughout the Genome of the Pacific Oyster Crassostrea Gigas Carried by Helentron Non-Autonomous Mobile Elements. Sci. Rep. 2020, 10, 15107. [Google Scholar] [CrossRef] [PubMed]
- Tunjić-Cvitanić, M.; Pasantes, J.J.; García-Souto, D.; Cvitanić, T.; Plohl, M.; Šatović-Vukšić, E. Satellitome Analysis of the Pacific Oyster Crassostrea Gigas Reveals New Pattern of Satellite Dna Organization, Highly Scattered across the Genome. Int. J. Mol. Sci. 2021, 22, 6798. [Google Scholar] [CrossRef]
- Dias, G.B.; Svartman, M.; Delprat, A.; Ruiz, A.; Kuhn, G.C.S. Tetris Is a Foldback Transposon That Provided the Building Blocks for an Emerging Satellite DNA of Drosophila Virilis. Genome Biol. Evol. 2014, 6, 1302–1313. [Google Scholar] [CrossRef]
- Kuhn, G.C.S.; Heringer, P.; Dias, G.B. Structure, Organization, and Evolution of Satellite DNAs: Insights from the Drosophila Repleta and D. Virilis Species Groups. Prog. Mol. Subcell. Biol. 2021, 60, 27–56. [Google Scholar] [CrossRef]
- Meštrović, N.; Mravinac, B.; Pavlek, M.; Vojvoda-Zeljko, T.; Šatović, E.; Plohl, M. Structural and Functional Liaisons between Transposable Elements and Satellite DNAs. Chromosom. Res. 2015, 23, 583–596. [Google Scholar] [CrossRef]
- Zattera, M.L.; Bruschi, D.P. Transposable Elements as a Source of Novel Repetitive DNA in the Eukaryote Genome. Cells 2022, 11, 3373. [Google Scholar] [CrossRef]
- Vondrak, T.; Avila Robledillo, L.; Novak, P.; Koblizkova, A.; Neumann, P.; Macas, J. Characterization of Repeat Arrays in Ultra-Long Nanopore Reads Reveals Frequent Origin of Satellite DNA from Retrotransposon-Derived Tandem Repeats. Plant J. 2020, 101, 484–500. [Google Scholar] [CrossRef]
- Brajković, J.; Feliciello, I.; Bruvo-Mađarić, B.; Ugarković, D. Satellite DNA-like Elements Associated with Genes within Euchromatin of the Beetle Tribolium Castaneum. G3 Genes Genomes Genet. 2012, 2, 931–941. [Google Scholar] [CrossRef]
- Kapitonov, V.V.; Gentles, A.J.; Jurka, J. Rehavkus-1_TC, a Family of Rehavkus DNA Transposons from the Red Flour Beetle Genome. Repbase Rep. 2006, 6, 193. [Google Scholar]
- Scalvenzi, T.; Pollet, N. Insights on Genome Size Evolution from a Miniature Inverted Repeat Transposon Driving a Satellite DNA. Mol. Phylogenet. Evol. 2014, 81, 1–9. [Google Scholar] [CrossRef]
- Pita, S.; Panzera, F.; Mora, P.; Vela, J.; Cuadrado, Á.; Sánchez, A.; Palomeque, T.; Lorite, P. Comparative Repeatome Analysis on Triatoma Infestans Andean and Non-Andean Lineages, Main Vector of Chagas Disease. PLoS ONE 2017, 12, e0181635. [Google Scholar] [CrossRef]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The Complete Sequence of a Human Genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SatDNA | Monomer Length (bp) | Genomic Abundance (%) | Chromosome | Original Name 1,2,3 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | Unplaced Scaffolds | Unplaced Singletons | ||||
| TCsat01 | 361 | 17 | □ | □ | □ | □ | □ | □ | □ | □ | □ | □ | ■ | TCAST 1 | |
| TCsat02 | 359 | 0.38 | □ | □ | □ | □ | □ | □ | □ | □ | □ | □ | ■ | TCAST2 2 | |
| TCsat03 | 172 | 1 | □ | ■ | ■ | ■ | ■ | ■ | ■ | ■ | ■ | ■ | □ | ■ | Cast1 3 |
| TCsat04 | 172 | 0.5 | ■ | ■ | ■ | ■ | ■ | ■ | □ | ■ | ■ | ■ | ■ | ■ | Cast2 3 |
| TCsat05 | 227 | 0.2 | ■ | ■ | ■ | ■ | ■ | ■ | ■ | ■ | ■ | ■ | □ | □ | Cast3 3 |
| TCsat06 | 179 | 0.5 | ■ | ■ | □ | ■ | ■ | ■ | ■ | ■ | □ | ■ | ■ | Cast4 3 | |
| TCsat07 | 334 | 1 | □ | ■ | ■ | ■ | ■ | ■ | ■ | ■ | ■ | ■ | ■ | ■ | Cast5 3 |
| TCsat08 | 180 | 0.5 | □ | ■ | ■ | ■ | □ | ■ | ■ | ■ | Cast6 3 | ||||
| TCsat09 | 121 | 0.2 | □ | □ | ■ | ■ | ■ | □ | □ | □ | □ | □ | □ | □ | Cast7 3 |
| TCsat10 | 169 | 0.2 | □ | □ | ■ | □ | □ | ■ | □ | ■ | ■ | ■ | ■ | ■ | Cast8 3 |
| TCsat11 | 350 | 0.2 | □ | □ | ■ | □ | ■ | □ | □ | □ | ■ | ■ | □ | Cast9 3 | |
| TCsat12 | 154 | 0.0461 | □ | ■ | ■ | ||||||||||
| TCsat13 | 266 | 0.0184 | ■ | ■ | |||||||||||
| TCsat14 | 309 | 0.0154 | □ | ■ | □ | ||||||||||
| TCsat15 | 1106 | 1.6857 | □ | □ | □ | ■ | □ | □ | □ | □ | □ | □ | □ | □ | |
| TCsat16 | 330 | 0.0059 | ■ | ||||||||||||
| TCsat17 | 120 | 0.0077 | ■ | ||||||||||||
| TCsat18 | 73 | 0.0892 | □ | □ | ■ | ■ | □ | □ | ■ | □ | □ | ■ | |||
| TCsat19 | 618 | 0.0174 | ■ | ■ | ■ | □ | |||||||||
| TCsat20 | 144 | 0.0045 | ■ | ||||||||||||
| TCsat21 | 102 | 0.0041 | ■ | ||||||||||||
| TCsat22 | 281 | 0.0040 | ■ | ||||||||||||
| TCsat23 | 164 | 0.0078 | ■ | ■ | ■ | □ | ■ | ||||||||
| TCsat24 | 721 | 0.0898 | □ | □ | ■ | ||||||||||
| TCsat25 | 258 | 0.0027 | ■ | ||||||||||||
| TCsat26 | 361 | 0.0030 | ■ | ■ | |||||||||||
| TCsat27 | 302 | 0.0040 | ■ | ■ | |||||||||||
| TCsat28 | 412 | 0.0036 | ■ | ||||||||||||
| TCsat29 | 174 | 0.0028 | □ | ■ | ■ | ||||||||||
| TCsat30 | 90 | 0.0019 | ■ | ||||||||||||
| TCsat31 | 144 | 0.0018 | ■ | ||||||||||||
| TCsat32 | 325 | 0.0033 | ■ | ||||||||||||
| TCsat33 | 150 | 0.0017 | ■ | ||||||||||||
| TCsat34 | 203 | 0.0020 | ■ | ||||||||||||
| TCsat35 | 172 | 0.0018 | ■ | ||||||||||||
| TCsat36 | 178 | 0.0020 | □ | ■ | |||||||||||
| TCsat37 | 230 | 0.0470 | □ | □ | □ | ■ | □ | ■ | ■ | □ | □ | □ | ■ | ||
| TCsat38 | 178 | 0.0070 | ■ | □ | □ | □ | ■ | □ | |||||||
| TCsat39 | 188 | 0.0018 | ■ | ||||||||||||
| TCsat40 | 276 | 0.0020 | ■ | ||||||||||||
| TCsat41 | 122 | 0.0016 | ■ | ||||||||||||
| TCsat42 | 179 | 0.0016 | □ | ■ | |||||||||||
| TCsat43 | 178 | 0.0016 | ■ | ||||||||||||
| TCsat44 | 311 | 0.0021 | ■ | ||||||||||||
| TCsat45 | 260 | 0.0017 | ■ | ||||||||||||
| TCsat46 | 142 | 0.0017 | ■ | ||||||||||||
| TCsat47 | 282 | 0.0019 | ■ | ||||||||||||
| TCsat48 | 156 | 0.0017 | ■ | ||||||||||||
| TCsat49 | 354 | 0.0017 | ■ | ||||||||||||
| TCsat50 | 168 | 0.0024 | ■ | ||||||||||||
| TCsat51 | 353 | 0.0043 | ■ | ||||||||||||
| TCsat52 | 184 | 0.0013 | ■ | ■ | |||||||||||
| TCsat53 | 152 | 0.0017 | ■ | ||||||||||||
| TCsat54 | 322 | 0.0022 | ■ | □ | |||||||||||
| TCsat55 | 311 | 0.0023 | ■ | ||||||||||||
| TCsat56 | 306 | 0.0024 | ■ | ||||||||||||
| TCsat57 | 212 | 0.0013 | ■ | ||||||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gržan, T.; Dombi, M.; Despot-Slade, E.; Veseljak, D.; Volarić, M.; Meštrović, N.; Plohl, M.; Mravinac, B. The Low-Copy-Number Satellite DNAs of the Model Beetle Tribolium castaneum. Genes 2023, 14, 999. https://doi.org/10.3390/genes14050999
Gržan T, Dombi M, Despot-Slade E, Veseljak D, Volarić M, Meštrović N, Plohl M, Mravinac B. The Low-Copy-Number Satellite DNAs of the Model Beetle Tribolium castaneum. Genes. 2023; 14(5):999. https://doi.org/10.3390/genes14050999
Chicago/Turabian StyleGržan, Tena, Mira Dombi, Evelin Despot-Slade, Damira Veseljak, Marin Volarić, Nevenka Meštrović, Miroslav Plohl, and Brankica Mravinac. 2023. "The Low-Copy-Number Satellite DNAs of the Model Beetle Tribolium castaneum" Genes 14, no. 5: 999. https://doi.org/10.3390/genes14050999