
JAK/STAT Signaling and Cervical Cancer: From the Cell Surface to the Nucleus

Abstract
1. Introduction
2. The Discovery of the JAK/STAT Pathway
3. The JAK/STAT Pathway
4. Players of the JAK/STAT Signaling Pathway
4.1. Cell Surface Receptors
4.2. JAKs
4.3. STATs
5. Negative Regulators
5.1. SOCS/CIS Family
5.2. PIAS
5.3. PTPs
6. Signaling Pathways and HPV
6.1. Interferon (IFN) Pathway
6.2. Wnt/β-Catenin Pathway
6.3. PI3K/AKT/mTOR Pathway
6.4. ERK/MAPK Pathway
6.5. Ying and Yang 1 (YY1) Pathway
6.6. EGFR Family Pathway
6.7. NF-κB Pathway
6.8. miRNAs
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Available online: https://gco.iarc.fr/today/data/factsheets/cancers/23-Cervix-uteri-fact-sheet.pdf (accessed on 10 March 2023).
- Arbyn, M.; Weiderpass, E.; Bruni, L.; de Sanjosé, S.; Saraiya, M.; Ferlay, J.; Bray, F. Estimates of incidence and mortality of cervical cancer in 2018: A worldwide analysis. Lancet Glob. Health 2020, 8, e191–e203. [Google Scholar] [CrossRef]
- Duranti, S.; Pietragalla, A.; Daniele, G.; Nero, C.; Ciccarone, F.; Scambia, G.; Lorusso, D. Role of immune checkpoint inhibitors in cervical cancer: From preclinical to clinical data. Cancers 2021, 13, 2089. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- de Sanjose, S.; Quint, W.G.V.; Alemany, L.; Geraets, D.T.; Klaustermeier, J.E.; Lloveras, B.; Tous, S.; Felix, A.; Bravo, L.E.; Shin, H.-R.; et al. Human papillomavirus genotype attribution in invasive cervical cancer: A retrospective cross-sectional worldwide study. Lancet Oncol. 2010, 11, 1048–1056. [Google Scholar] [CrossRef]
- Songock, W.K.; Kim, S.-M.; Bodily, J.M. The human papillomavirus E7 oncoprotein as a regulator of transcription. Virus Res. 2017, 231, 56–75. [Google Scholar] [CrossRef]
- Yeo-Teh, N.S.L.; Ito, Y.; Jha, S. High-Risk Human Papillomaviral Oncogenes E6 and E7 Target Key Cellular Pathways to Achieve Oncogenesis. Int. J. Mol. Sci. 2018, 19, 1706. [Google Scholar] [CrossRef]
- Sharma, S.; Munger, K. The role of Long Noncoding RNAs in Human Papillomavirus-Associated Phatogenesis. Phatogens 2020, 9, 289. [Google Scholar]
- Zheng, Z.-M.; Wang, X. Regulation of cellular miRNA expression by human papillomaviruses. Biochim. Biophys. Acta Gene Regul. Mech. 2011, 1809, 668–677. [Google Scholar] [CrossRef]
- Carter, J.R.; Ding, Z.; Rose, B.R. HPV infection and cervical disease: A review. Aust. N. Zldn. J. Obstet. Gynaecol. 2011, 51, 103–108. [Google Scholar] [CrossRef]
- Moerman-Herzog, A.; Nakagawa, M. Early Defensive Mechanisms against Human Papillomavirus Infection. Clin. Vaccine Immunol. 2015, 22, 850–857. [Google Scholar] [CrossRef]
- Muñoz, N.; Bosch, F.X.; De Sanjosé, S.; Herrero, R.; Castellsagué, X.; Shah, K.V.; Snijders, P.J.F.; Meijer, C.J.L.M. Epidemiologic Classification of Human Papillomavirus Types Associated with Cervical Cancer. N. Engl. J. Med. 2003, 348, 518–527. [Google Scholar] [CrossRef]
- Gillet, E.; Meys, J.F.; Verstraelen, H.; Bosire, C.; De Sutter, P.; Temmerman, M.; Broeck, D.V. Bacterial vaginosis is associated with uterine cervical human papillomavirus infection: A meta-analysis. BMC Infect. Dis. 2011, 11, 10. [Google Scholar] [CrossRef]
- Guo, Y.-L.; You, K.; Qiao, J.; Zhao, Y.-M.; Geng, L. Bacterial vaginosis is conducive to the persistence of HPV infection. Int. J. STD AIDS 2012, 23, 581–584. [Google Scholar] [CrossRef]
- Vriend, H.J.; Bogaards, J.A.; van Bergen, J.E.A.M.; Brink, A.A.T.P.; van den Broek, I.V.F.; Hoebe, C.J.P.A.; King, A.J.; van der Sande, M.A.B.; Wolffs, P.F.G.; de Melker, H.E.; et al. Incidence and persistence of carcinogenic genital human papillomavirus infections in young women with or without Chlamydia trachomatis co-infection. Cancer Med. 2015, 4, 1589–1598. [Google Scholar] [CrossRef]
- Clarke, M.A.; Rodriguez, A.C.; Gage, J.C.; Herrero, R.; Hildesheim, A.; Wacholder, S.; Burk, R.; Schiffman, M. A large, population-based study of age-related associations between vaginal pH and human papillomavirus infection. BMC Infect. Dis. 2012, 12, 33. [Google Scholar] [CrossRef]
- Gutiérrez-Hoya, A.; Soto-Cruz, I. Role of the JAK/STAT Pathway in Cervical Cancer: Its Relationship with HPV E6/E7 Oncoproteins. Cells 2020, 9, 2297. [Google Scholar] [CrossRef]
- Morgan, E.L.; Macdonald, A. Manipulation of JAK/STAT Signalling by High-Risk HPVs: Potential Therapeutic Targets for HPV-Associated Malignancies. Viruses 2020, 12, 977. [Google Scholar] [CrossRef]
- Owen, K.L.; Brockwell, N.K.; Parker, B.S. JAK-STAT Signaling: A Double-Edged Sword of Immune Regulation and Cancer Progression. Cancers 2019, 11, 2002. [Google Scholar] [CrossRef]
- Harrison, D.A. The Jak/STAT pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011205. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr. STATs and gene regulation. Science 1997, 277, 1630–1635. [Google Scholar] [CrossRef]
- Stark, G.R.; Darnell, J.E., Jr. The JAK-STAT Pathway at Twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef]
- Oashea, J.; O’shea, J.J.; Husa, M.; Li, D.; Hofmann, S.R.; Watford, W.; Roberts, J.L.; Buckley, R.H.; Changelian, P.; Candotti, F. Jak3 and the pathogenesis of severe combined immunodeficiency. Mol. Immunol. 2004, 41, 727–737. [Google Scholar] [CrossRef]
- Firmbach-Kraft, I.; Byers, M.; Shows, T.; Dalla-Favera, R.; Krolewski, J.J. tyk2, prototype of a novel class of non-receptor tyrosine kinase genes. Oncogene 1990, 5, 1329–1336. [Google Scholar]
- Sandberg, E.M.; Wallace, T.A.; Godeny, M.D.; Linden, D.V.; Sayeski, P.P. Jak2 tyrosine kinase: A true jak of all trades? Cell Biochem. Biophys. 2004, 41, 207–232. [Google Scholar] [CrossRef]
- Rane, S.G.; Reddy, E.P. Janus kinases: Components of multiple signaling pathways. Oncogene 2000, 19, 5662–5679. [Google Scholar] [CrossRef]
- Harpur, A.G.; Andres, A.C.; Ziemiecki, A.; Aston, R.R.; Wilks, A.F. JAK2, a third member of the JAK family of protein tyrosine kinases. Oncogene 1992, 7, 1347–1353. [Google Scholar]
- Shuai, K.; Schindler, C.; Prezioso, V.R.; Darnell, J.E., Jr. Activation of transcription by IFN-gamma: Tyrosine phosphorylation of a 91-kD DNA binding protein. Science 1992, 258, 1808–1812. [Google Scholar] [CrossRef]
- Shuai, K.; Stark, G.R.; Kerr, I.M.; Darnell, J.E., Jr. A single phosphotyrosine residue of Stat91 required for gene activation by interferon-gamma. Science 1993, 261, 1744–1746. [Google Scholar] [CrossRef]
- Zhong, Z.; Wen, Z.; Darnell, J.E., Jr. Stat3: A STAT Family Member Activated by Tyrosine Phosphorylation in Response to Epidermal Growth Factor and Interleukin-6. Science 1994, 264, 95–98. [Google Scholar] [CrossRef]
- Zhong, Z.; Wen, Z.; E Darnell, J. Stat3 and Stat4: Members of the family of signal transducers and activators of transcription. Proc. Natl. Acad. Sci. USA 1994, 91, 4806–4810. [Google Scholar] [CrossRef]
- Wakao, H.; Gouilleux, F.; Groner, B. Mammary gland factor (MGF) is a novel member of the cytokine regulated transcription factor gene family and confers the prolactin response. EMBO J. 1994, 13, 2182–2191. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, L.; Fellous, M.; Stark, G.R.; Pellegrini, S. A protein tyrosine kinase in the interferon alpha/beta signaling pathway. Cell 1992, 70, 313–322. [Google Scholar] [CrossRef]
- Müller, M.; Briscoe, J.; Laxton, C.; Guschin, D.; Ziemiecki, A.; Silvennoinen, O.; Harpur, A.G.; Barbieri, G.; Witthuhn, B.A.; Schindler, C.; et al. The protein tyrosine kinase JAK1 complements defects in interferon-alpha/beta and -gamma signal transduction. Nature 1993, 366, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Tenhumberg, S.; Schuster, B.; Zhu, L.; Kovaleva, M.; Scheller, J.; Kallen, K.-J.; Rose-John, S. gp130 dimerization in the absence of ligand: Preformed cytokine receptor complexes. Biochem. Biophys. Res. Commun. 2006, 346, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Livnah, O.; Stura, E.A.; Middleton, S.A.; Johnson, D.L.; Jolliffe, L.K.; Wilson, I.A. Crystallographic Evidence for Preformed Dimers of Erythropoietin Receptor Before Ligand Activation. Science 1999, 283, 987–990. [Google Scholar] [CrossRef] [PubMed]
- Remy, I.; Wilson, I.A.; Michnick, S.W. Erythropoietin Receptor Activation by a Ligand-Induced Conformation Change. Science 1999, 283, 990–993. [Google Scholar] [CrossRef] [PubMed]
- Naismith, J.H.; Devine, T.Q.; Brandhuber, B.J.; Sprang, S.R. Crystallographic evidence for dimerization of unliganded tumor necrosis factor receptor. J. Biol. Chem. 1995, 270, 13303–13307. [Google Scholar] [CrossRef] [PubMed]
- Kramer, J.M.; Yi, L.; Shen, F.; Maitra, A.; Jiao, X.; Jin, T.; Gaffen, S.L. Cutting Edge: Evidence for Ligand-Independent Multimerization of the IL-17 Receptor. J. Immunol. 2006, 176, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Krause, C.D.; Mei, E.; Mirochnitchenko, O.; Lavnikova, N.; Xie, J.; Jia, Y.; Hochstrasser, R.M.; Pestka, S. Interactions among the components of the interleukin-10 receptor complex. Biochem. Biophys. Res. Commun. 2006, 340, 377–385. [Google Scholar] [CrossRef]
- Brooks, A.J.; Dai, W.; O’mara, M.L.; Abankwa, D.; Chhabra, Y.; Pelekanos, R.A.; Gardon, O.; Tunny, K.A.; Blucher, K.M.; Morton, C.J.; et al. Mechanism of Activation of Protein Kinase JAK2 by the Growth Hormone Receptor. Science 2014, 344, 1249783. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.B.; Laurence, A. The JAK-STAT pathway: Impact on human disease and therapeutic intervention. Annu. Rev. Med. 2015, 66, 311–328. [Google Scholar] [CrossRef] [PubMed]
- Durham, G.; Williams, J.J.; Nasim, T.; Palmer, T.M. Targeting SOCS Proteins to Control JAK-STAT Signalling in Disease. Trends Pharmacol. Sci. 2019, 40, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Bourke, L.T.; Knight, R.A.; Latchman, D.S.; Stephanou, A.; McCormick, J. Signal transducer and activator of transcription-1 localizes to the mitochondria and modulates mitophagy. JAK-STAT 2013, 2, e25666. [Google Scholar] [CrossRef]
- Sisler, J.D.; Morgan, M.; Raje, V.; Grande, R.C.; Derecka, M.; Meier, J.; Cantwell, M.; Szczepanek, K.; Korzun, W.J.; Lesnefsky, E.J.; et al. The Signal Transducer and Activator of Transcription 1 (STAT1) Inhibits Mitochondrial Biogenesis in Liver and Fatty Acid Oxidation in Adipocytes. PLoS ONE 2015, 10, e0144444. [Google Scholar] [CrossRef] [PubMed]
- Shahni, R.; Cale, C.M.; Anderson, G.; Osellame, L.D.; Hambleton, S.; Jacques, T.S.; Wedatilake, Y.; Taanman, J.-W.; Chan, E.; Qasim, W.; et al. Signal transducer and activator of transcription 2 deficiency is a novel disorder of mitochondrial fission. Brain 2015, 138, 2834–2846. [Google Scholar] [CrossRef]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef]
- Wegrzyn, J.; Potla, R.; Chwae, Y.-J.; Sepuri, N.B.V.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of Mitochondrial Stat3 in Cellular Respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef]
- Szczepanek, K.; Lesnefsky, E.J.; Larner, A.C. Multi-tasking: Nuclear transcription factors with novel roles in the mitochondria. Trends Cell Biol. 2012, 22, 429–437. [Google Scholar] [CrossRef]
- Sehgal, P.B. Non-genomic STAT5-dependent effects at the endoplasmic reticulum and Golgi apparatus and STAT6-GFP in mitochondria. JAK-STAT 2013, 2, e24860. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Yang, Y.-M.; Liang, F.-X.; Gough, D.J.; Levy, D.; Sehgal, P.B. Nongenomic STAT5-dependent effects on Golgi apparatus and endoplasmic reticulum structure and function. Am. J. Physiol. Cell Physiol. 2012, 302, C804–C820. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, A.; Shishodia, S.; Fu, X.Y.; Aggarwal, B. Lack of requierement of STAT1 for activation of nuclear factor-kappaB, c-Jun NH2-terminal protein kinase, and apoptosis by tumor necrosis factor-alpha. J. Cell. Biochem. 2002, 84, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.; Zeidler, M.P. Unphosphorylated STATs go nuclear. Curr. Opin. Genet. Dev. 2008, 18, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Larson, K.; Guo, D.; Lim, S.J.; Dutta, P.; Yan, S.-J.; Li, W.X. Drosophila STAT is required for directly maintaining HP1 localization and heterochromatin stability. Nat. Cell Biol. 2008, 10, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Li, W.X. Canonical and non-canonical JAK–STAT signaling. Trends Cell Biol. 2008, 18, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Calhoun, H.C.; Xia, F.; Li, J.; Le, L.; Li, W.X. JAK signaling globally counteracts heterochromatic gene silencing. Nat. Genet. 2006, 38, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Dutta, P.; Tsurumi, A.; Li, J.; Wang, J.; Land, H.; Li, W.X. Unphosphorylated STAT5A stabilizes heterochromatin and suppresses tumor growth. Proc. Natl. Acad. Sci. USA 2013, 110, 10213–10218. [Google Scholar] [CrossRef]
- Park, H.J.; Li, J.; Hannah, R.; Biddie, S.; Leal-Cervantes, A.I.; Kirschner, K.; Cruz, D.F.S.; Sexl, V.; Göttgens, B.; Green, A.R. Cytokine-induced megakaryocytic differentiation is regulated by genome-wide loss of a uSTAT transcriptional program. EMBO J. 2015, 35, 580–594. [Google Scholar] [CrossRef]
- Yang, J.; Liao, X.; Agarwal, M.K.; Barnes, L.; Auron, P.E.; Stark, G.R. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFκB. Genes Dev. 2007, 21, 1396–1408. [Google Scholar] [CrossRef]
- Cui, X.; Zhang, L.; Luo, J.; Rajasekaran, A.; Hazra, S.; Cacalano, N.; Dubinett, S.M. Unphosphorylated STAT6 contributes to constitutive cyclooxygenase-2 expression in human non-small cell lung cancer. Oncogene 2007, 26, 4253–4260. [Google Scholar] [CrossRef]
- Chatterjee-Kishore, M.; Wright, K.L.; Ting, J.P.-Y.; Stark, G.R. How Stat1 mediates constitutive gene expression: A complex of unphosphorylated Stat1 and IRF1 supports transcription of the LMP2 gene. EMBO J. 2000, 19, 4111–4122. [Google Scholar] [CrossRef]
- Meyer, T.; Gavenis, K.; Vinkemeier, U. Cell Type-Specific and Tyrosine Phosphorylation-Independent Nuclear Presence of STAT1 and STAT3. Exp. Cell Res. 2002, 272, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Marg, A.; Shan, Y.; Meyer, T.; Meissner, T.; Brandenburg, M.; Vinkemeier, U. Nucleocytoplasmic shuttling by nucleoporins Nup153 and Nup214 and CRM1-dependent nuclear export control the subcellular distribution of latent Stat1. J. Cell Biol. 2004, 165, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.Y.; Johnson, F.M. Defining the role of the JAK-STAT pathway in head and neck and thoracic malignancies: Implications for future therapeutic approaches. Drug Resist. Updates 2010, 13, 67–78. [Google Scholar] [CrossRef]
- Limnander, A.; Rothman, P.B. Abl Oncogene Bypasses Normal Regulation of JAK/STAT Activation. Cell Cycle 2004, 3, 1486–1488. [Google Scholar] [CrossRef]
- Coppo, P.; Flamant, S.; De Mas, V.; Jarrier, P.; Guillier, M.; Bonnet, M.-L.; Lacout, C.; Guilhot, F.; Vainchenker, W.; Turhan, A.G. BCR-ABL activates STAT3 via JAK and MEK pathways in human cells. Br. J. Haematol. 2006, 134, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.L.; Meyer, D.J.; Campbell, G.S.; Larner, A.C.; Carter-Su, C.; Schwartz, J.; Jove, R. Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science 1995, 269, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Ruff-Jamison, S.; Zhong, Z.; Wen, Z.; Chen, K.; Darnell, J.; Cohen, S. Epidermal growth factor and lipopolysaccharide activate Stat3 transcription factor in mouse liver. J. Biol. Chem. 1994, 269, 21933–21935. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.Y.; Zhang, J.J. Transcription factor p91 interacts with the epidermal growth factor receptor and mediates activation of the c-fos gene promoter. Cell 1993, 74, 1135–1145. [Google Scholar] [CrossRef]
- Ruff-Jamison, S.; Chen, K.; Cohen, S. Epidermal growth factor induces the tyrosine phosphorylation and nuclear translocation of Stat 5 in mouse liver. Proc. Natl. Acad. Sci. USA 1995, 92, 4215–4218. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Pardanani, A.; Tefferi, A.; Gilliland, D.G. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat. Rev. Cancer 2007, 7, 673–683. [Google Scholar] [CrossRef]
- Tang, L.-Y.; Heller, M.; Meng, Z.; Yu, L.-R.; Tang, Y.; Zhou, M.; Zhang, Y.E. Transforming Growth Factor-β (TGF-β) Directly Activates the JAK1-STAT3 Axis to Induce Hepatic Fibrosis in Coordination with the SMAD Pathway. J. Biol. Chem. 2017, 292, 4302–4312. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Yu, Y.; Sun, C.; Liu, T.; Liang, T.; Zhan, L.; Lin, X.; Feng, X.-H. STAT3 selectively interacts with Smad3 to antagonize TGF-β signalling. Oncogene 2016, 35, 4388–4398. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Herrmann, A.; Deng, J.H.; Kujawski, M.; Niu, G.; Li, Z.; Forman, S.; Jove, R.; Pardoll, D.M.; Yu, H. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell 2009, 15, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Bousoik, E.; Aliabadi, H.M. “Do We Know Jack” About JAK? A Closer Look at JAK/STAT Signaling Pathway. Front. Oncol. 2018, 8, 287. [Google Scholar] [CrossRef]
- Liao, W.; Lin, J.-X.; Leonard, W.J. Interleukin-2 at the Crossroads of Effector Responses, Tolerance, and Immunotherapy. Immunity 2013, 38, 13–25. [Google Scholar] [CrossRef]
- Rocha-Zavaleta, L.; Huitron, C.; Cacéres-Cortés, J.R.; Alvarado-Moreno, J.A.; Valle-Mendiola, A.; Soto-Cruz, I.; Weiss-Steider, B.; Rangel-Corona, R. Interleukin-2 (IL-2) receptor-βγ signalling is activated by c-Kit in the absence of IL-2, or by exogenous IL-2 via JAK3/STAT5 in human papillomavirus-associated cervical cancer. Cell. Signal. 2004, 16, 1239–1247. [Google Scholar] [CrossRef]
- Valle-Mendiola, A.; Weiss-Steider, B.; Rocha-Zavaleta, L.; Soto-Cruz, I. IL-2 Enhances Cervical Cancer Cells Proliferation and JAK3/STAT5 Phosphorylation at Low Doses, While at High Doses IL-2 Has Opposite Effects. Cancer Investig. 2014, 32, 115–125. [Google Scholar] [CrossRef]
- Friedmann, M.C.; Migone, T.S.; Russell, S.M.; Leonard, W.J. Different interleukin 2 receptor beta-chain tyrosines couple to at least two signaling pathways and synergistically mediate interleukin 2-induced proliferation. Proc. Natl. Acad. Sci. USA 1996, 93, 2077–2082. [Google Scholar] [CrossRef]
- García-Tuñón, I.; Ricote, M.; Ruiz, A.; Fraile, B.; Paniagua, R.; Royuela, M. Interleukin-2 and its receptor complex (α, β and γ chains) in in situand infiltrative human breast cancer: An immunohistochemical comparative study. Breast Cancer Res. 2003, 6, R1–R7. [Google Scholar] [CrossRef]
- Lagunas-Cruz, M.D.C.; Valle-Mendiola, A.; Trejo-Huerta, J.; Rocha-Zavaleta, L.; Mora-García, M.D.L.; Gutiérrez-Hoya, A.; Weiss-Steider, B.; Soto-Cruz, I. IL-2 Induces Transient Arrest in the G1 Phase to Protect Cervical Cancer Cells from Entering Apoptosis. J. Oncol. 2019, 2019, 7475295. [Google Scholar] [CrossRef]
- D’Andrea, A.D.; Fasman, G.D.; Lodish, H.F. Erythropoietin receptor and interleukin-2 receptor beta chain: A new receptor family. Cell 1989, 58, 1023–1024. [Google Scholar] [CrossRef] [PubMed]
- Barber, D.L.; D’Andrea, A.D. Erythropoietin and Interleukin-2 Activate Distinct JAK Kinase Family Members. Mol. Cell. Biol. 1994, 14, 6506–6514. [Google Scholar] [PubMed]
- Lopez, T.V.; Lappin, T.R.; Maxwell, P.; Shi, Z.; Lopez-Marure, R.; Aguilar, C.; Rocha-Zavaleta, L. Autocrine/paracrine erythropoietin signalling promotes JAK/STAT-dependent proliferation of human cervical cancer cells. Int. J. Cancer 2011, 129, 2566–2576. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. The JAK-STAT Signaling Pathway: Input and Output Integration. J. Immunol. 2007, 178, 2623–2629. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Bhattacharjee, A.; Xu, B.; Ford, D.; Maizel, A.L.; Cathcart, M.K. IL-13 signal transduction in human monocytes: Phosphorylation of receptor components, association with Jaks, and phosphorylation/activation of Stats. J. Leukoc. Biol. 2002, 72, 580–589. [Google Scholar] [CrossRef]
- Chen, Z.; Lund, R.; Aittokallio, T.; Kosonen, M.; Nevalainen, O.; Lahesmaa, R. Identification of Novel IL-4/Stat6-Regulated Genes in T Lymphocytes. J. Immunol. 2003, 171, 3627–3635. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.A.; Siddiqui, M. The JAK-STAT pathway in hypertrophic stress signaling and genomic stress response. JAK-STAT 2012, 1, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Vila-Coro, A.J.; Rodríguez-Frade, J.M.; de Ana, A.M.; Moreno-Ortíz, M.C.; Martínez, A.C.; Mellado, M. The chemokine SDF-1alpha triggers CXCR4 receptor dimerization and activates the JAK/STAT pathway. FASEB J. 1999, 13, 1699–1710. [Google Scholar] [CrossRef] [PubMed]
- Andl, C.D.; Mizushima, T.; Oyama, K.; Bowser, M.; Nakagawa, H.; Rustgi, A.K. EGFR-induced cell migration is mediated predominantly by the JAK-STAT pathway in primary esophageal keratinocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G1227–G1237. [Google Scholar] [CrossRef]
- Raju, R.; Palapetta, S.M.; Sandhya, V.K.; Sahu, A.; Alipoor, A.; Balakrishnan, L.; Advani, J.; George, B.; Kini, K.R.; Geetha, N.P.; et al. A Network Map of FGF-1/FGFR Signaling System. J. Signal Transduct. 2014, 2014, 962962. [Google Scholar] [CrossRef]
- Dudka, A.A.; Sweet, S.M.; Heath, J.K. Signal transducers and activators of transcription-3 binding to the fibroblast growth factor receptor is activated by receptor amplification. Cancer Res. 2010, 70, 3391–3401. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Pan, C.; Sun, J.; Gilbert, C.; Drews-Elger, K.; Azzam, D.J.; Picon-Ruiz, M.; Kim, M.; Ullmer, W.; El-Ashry, D.; et al. VEGF drives cancer-initiating stem cells through VEGFR-2/Stat3 signaling to upregulate Myc and Sox2. Oncogene 2014, 34, 3107–3119. [Google Scholar] [CrossRef]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Eyking, A.; Ey, B.; Rünzi, M.; Roig, A.I.; Reis, H.; Schmid, K.W.; Gerken, G.; Podolsky, D.K.; Cario, E. Toll-like Receptor 4 Variant D299G Induces Features of Neoplastic Progression in Caco-2 Intestinal Cells and Is Associated with Advanced Human Colon Cancer. Gastroenterology 2011, 141, 2154–2165. [Google Scholar] [CrossRef]
- Liu, C.; Gao, F.; Li, B.; Mitchel, R.E.J.; Liu, X.; Lin, J.; Zhao, L.; Cai, J. TLR4 knockout protects mice from radiation-induced thymic lymphoma by downregulation of IL6 and miR-21. Leukemia 2011, 25, 1516–1519. [Google Scholar] [CrossRef]
- Herrmann, A.; Cherryholmes, G.; Schroeder, A.; Phallen, J.; Alizadeh, D.; Xin, H.; Wang, T.; Lee, H.; Lahtz, C.; Swiderski, P.; et al. TLR9 Is Critical for Glioma Stem Cell Maintenance and Targeting. Cancer Res. 2014, 74, 5218–5228. [Google Scholar] [CrossRef] [PubMed]
- Wilks, A.F.; Harpur, A.G.; Kurban, R.R.; Ralph, S.J.; Zurcher, G.; Ziemiecki, A. Two novel protein-tyrosine kinases, each with a second phosphotransferase-related catalytic domain, define a new class of protein kinase. Mol. Cell. Biol. 1991, 11, 2057–2065. [Google Scholar] [PubMed]
- Ferrao, R.; Lupardus, P.J. The Janus Kinase (JAK) FERM and SH2 Domains: Bringing Specificity to JAK–Receptor Interactions. Front. Endocrinol. 2017, 8, 71. [Google Scholar] [CrossRef]
- Frank, S.J.; Yi, W.; Zhao, Y.; Goldsmith, J.F.; Gilliland, G.; Jiang, J.; Sakai, I.; Kraft, A.S. Regions of the JAK2 Tyrosine Kinase Required for Coupling to the Growth Hormone Receptor. J. Biol. Chem. 1995, 270, 14776–14785. [Google Scholar] [CrossRef]
- Velazquez, L.; Mogensen, K.E.; Barbieri, G.; Fellous, M.; Uzé, G.; Pellegrini, S. Distinct domains of the protein tyrosine kinase tyk2 required for binding of interferon-alpha/beta and for signal transduction. J. Biol. Chem. 1995, 270, 3327–3334. [Google Scholar] [CrossRef]
- Leonard, W.J.; O’Shea, J.J. JAKS AND STATS: Biological Implications. Annu. Rev. Immunol. 1998, 16, 293–322. [Google Scholar] [CrossRef] [PubMed]
- Rodig, S.J.; Meraz, M.A.; White, J.; Lampe, P.A.; Riley, J.K.; Arthur, C.D.; King, K.L.; Sheehan, K.C.; Yin, L.; Pennica, D.; et al. Disruption of the Jak1 Gene Demonstrates Obligatory and Nonredundant Roles of the Jaks in Cytokine-Induced Biologic Responses. Cell 1998, 93, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef] [PubMed]
- Schindler, C.; Strehlow, I. Cytokines and STAT signaling. Adv. Pharm. 2000, 47, 113–174. [Google Scholar]
- Russell, S.M.; Johnston, J.A.; Noguchi, M.; Kawamura, M.; Bacon, C.M.; Friedmann, M.; Berg, M.; McVicar, D.W.; Witthuhn, B.A.; Silvennoinen, O.; et al. Interaction of IL-2R beta and gamma c chains with Jak1 and Jak3: Implications for XSCID and XCID. Science 1994, 266, 1042–1045. [Google Scholar] [CrossRef]
- Gauzzi, M.C.; Velazquez, L.; McKendry, R.; Mogensen, K.E.; Fellous, M.; Pellegrini, S. Interferon-alpha-dependent activation of Tyk2 requieres phosphorylation of positive regulatory tyrosines by another kinase. J. Biol. Chem. 1996, 271, 20494–20500. [Google Scholar] [CrossRef]
- Stahl, N.; Boulton, T.G.; Farruggella, T.; Ip, N.Y.; Davis, S.; Witthuhn, B.A.; Quelle, F.W.; Silvennoinen, O.; Barbieri, G.; Pellegrini, S.; et al. Association and activation of Jak-Tyk kinases by CNTF-LIF-OSM-IL-6 beta receptor components. Science 1994, 263, 92–95. [Google Scholar] [CrossRef]
- Finbloom, D.S.; Winestock, K.D. IL-10 induces the tyrosine phosphorylation of tyk2 and Jak1 and the differential assembly of STAT1 alpha and STAT3 complexes in human T cells and monocytes. J. Immunol. 1995, 155, 1079–1090. [Google Scholar] [CrossRef]
- Bacon, C.; McVicar, D.W.; Ortaldo, J.R.; Rees, R.C.; O’Shea, J.J.; Johnston, J.A. Interleukin 12 (IL-12) induces tyrosine phosphorylation of JAK2 and TYK2: Differential use of Janus family tyrosine kinases by IL-2 and IL-12. J. Exp. Med. 1995, 181, 399–404. [Google Scholar] [CrossRef]
- Welham, M.J.; Learmonth, L.; Bone, H.; Schrader, J.W. Interleukin-13 signal transduction in lymphohemopoietic cells: Similarities and differences in signal transduction with interleukin-4 and insulin. J. Biol. Chem. 1995, 270, 12286–12296. [Google Scholar] [CrossRef]
- Fu, X.-Y. A transcription factor with SH2 and SH3 domains is directly activated by an interferon α-induced cytoplasmic protein tyrosine kinase(s). Cell 1992, 70, 323–335. [Google Scholar] [CrossRef]
- Hou, J.; Schindler, U.; Henzel, W.J.; Ho, T.C.; Brasseur, M.; McKnight, S.L. An Interleukin-4-Induced Transcription Factor: IL-4 Stat. Science 1994, 265, 1701–1706. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.Y.; Schindler, C.; Improta, T.; Aebersold, R.; Darnell, J.E., Jr. The proteins of ISGF-3, the interferon alpha-induced transcriptional activator, define a gene family involved in signal transduction. Proc. Natl. Acad. Sci. USA 1992, 89, 7840–7843. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Schindler, U.; Henzel, W.J.; Wong, S.C.; McKnight, S.L. Identification and purification of human stat proteins activated in response to interleukin-2. Immunity 1995, 2, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Mui, A.; Wakao, H.; O’Farrell, A.; Harada, N.; Miyajima, A. Interleukin-3, granulocyte-macrophage colony stimulating factor and interleukin-5 transduce signals through two STAT5 homologs. EMBO J. 1995, 14, 1166–1175. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, F.; Qadree, A.K.; Radu, T.B.; Orlova, A.; de Araujo, E.D.; Israelian, J.; Valent, P.; Mustjoki, S.M.; Herling, M.; Moriggl, R.; et al. Structural and mutational analysis of member-specific STAT functions. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2022, 1866, 130058. [Google Scholar] [CrossRef] [PubMed]
- Horvath, C.M. STAT proteins and transcriptional responses to extracellular signals. Trends Biochem. Sci. 2000, 25, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.L.; Geissal, E.D.; Farrar, J.D.; Murphy, K.M. Role of the Stat4 N Domain in Receptor Proximal Tyrosine Phosphorylation. Mol. Cell. Biol. 2000, 20, 7121–7131. [Google Scholar] [CrossRef]
- Shuai, K. Modulation of STAT signaling by STAT-interacting proteins. Oncogene 2000, 19, 2638–2644. [Google Scholar] [CrossRef] [PubMed]
- Vinkemeier, U.; Moarefi, I.; Darnell, J.E., Jr.; Kuriyan, J. Structure of the Amino-Terminal Protein Interaction Domain of STAT-4. Science 1998, 279, 1048–1052. [Google Scholar] [CrossRef]
- Begitt, A.; Meyer, T.; van Rossum, M.; Vinkemeier, U. Nucleocytoplasmic translocation of Stat1 is regulated by a leucine-rich export signal in the coiled-coil domain. Proc. Natl. Acad. Sci. USA 2000, 97, 10418–10423. [Google Scholar] [CrossRef] [PubMed]
- Collum, R.G.; Brutsaert, S.; Lee, G.; Schindler, C. A Stat3-interacting protein (StIP1) regulates cytokine signal transduction. Proc. Natl. Acad. Sci. USA 2000, 97, 10120–10125. [Google Scholar] [CrossRef]
- Horvath, C.M.; Stark, G.R.; Kerr, I.M.; Darnell, J.E., Jr. Interactions between STAT and non-STAT proteins in the interferon-stimulated gene factor 3 transcription complex. Mol. Cell. Biol. 1996, 16, 6957–6964. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Bhattacharya, S.; Braunstein, J.; Schindler, C. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene 2002, 285, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-G.; Farley, A.; Nicholson, S.E.; Willson, T.A.; Zugaro, L.M.; Simpson, R.J.; Moritz, R.L.; Cary, D.; Richardson, R.; Hausmann, G.; et al. The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc. Natl. Acad. Sci. USA 1999, 96, 2071–2076. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Kee, W.H.; Seow, K.T.; Fung, W.; Cao, X. The Coiled-Coil Domain of Stat3 Is Essential for Its SH2 Domain-Mediated Receptor Binding and Subsequent Activation Induced by Epidermal Growth Factor and Interleukin-6. Mol. Cell. Biol. 2000, 20, 7132–7139. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; John, S.; Berg, M.; Leonard, W.J. Functional association of Nmi with Stat5 and Stat1 in IL-2- and IFNgamma-mediated signaling. Cell 1999, 96, 121–130. [Google Scholar] [CrossRef]
- Ginter, T.; Fahrer, J.; Kröhnert, U.; Fetz, V.; Garrone, A.; Stauber, R.H.; Reichardt, W.; Müller-Newen, G.; Kosan, C.; Heinzel, T.; et al. Arginine residues within the DNA binding domain of STAT3 promote intracellular shuttling and phosphorylation os STAT3. Cell. Signal. 2014, 26, 1698–1706. [Google Scholar] [CrossRef]
- McBride, K.M.; McDonald, C.; Reich, N.C. Nuclear export signal located within theDNA-binding domain of the STAT1transcription factor. EMBO J. 2002, 19, 6196–6206. [Google Scholar] [CrossRef]
- Bromberg, J.; Chen, X. STAT proteins: Signal transducers amd activators of transcription. Methods Enzym. 2001, 333, 138–151. [Google Scholar]
- Yang, E.; Wen, Z.; Haspel, R.L.; Zhang, J.J.; Darnell, J.E., Jr. The Linker Domain of Stat1 Is Required for Gamma Interferon-Driven Transcription. Mol. Cell. Biol. 1999, 19, 5106–5112. [Google Scholar] [CrossRef] [PubMed]
- Kawata, T.; Shevchenko, A.; Fukuzawa, M.; Jermyn, K.A.; Totty, N.F.; Zhukovskaya, N.V.; Sterling, A.E.; Mann, M.; Williams, J.G. SH2 Signaling in a Lower Eukaryote: A STAT Protein That Regulates Stalk Cell Differentiation in Dictyostelium. Cell 1997, 89, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Barahmand-Pour, F.; Meinke, A.; Groner, B.; Decker, T. Jak2-Stat5 Interactions Analyzed in Yeast. J. Biol. Chem. 1998, 273, 12567–12575. [Google Scholar] [CrossRef]
- Chen, X.; Vinkemeier, U.; Zhao, Y.; Jeruzalmi, D.; Darnell, J.E.; Kuriyan, J. Crystal Structure of a Tyrosine Phosphorylated STAT-1 Dimer Bound to DNA. Cell 1998, 93, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Yan, H.; Wong, L.H.; Ralph, S.; Krolewski, J.; Schindler, C. The SH2 domains of Stat1 and Stat2 mediate multiple interactions in the transduction of IFN-alpha signals. EMBO J. 1996, 15, 1075–1084. [Google Scholar] [CrossRef]
- Shuai, K.; Horvath, C.M.; Huang, L.H.; Qureshi, S.A.; Cowburn, D.; Darnell, J.E. Interferon activation of the transcription factor Stat91 involves dimerization through SH2-phosphotyrosyl peptide interactions. Cell 1994, 76, 821–828. [Google Scholar] [CrossRef]
- Orlova, A.; Wagner, C.; de Araujo, E.D.; Bajusz, D.; Neubauer, H.A.; Herling, M.; Gunning, P.T.; Keserű, G.M.; Moriggl, R. Direct Targeting Options for STAT3 and STAT5 in Cancer. Cancers 2019, 11, 1930. [Google Scholar] [CrossRef]
- Yan, R.; Qureshi, S.; Zhong, Z.; Wen, Z.; Darnell, J.E. The genomic structure of the STAT genes: Multiple exons in coincident sites in Stat1 and Stat2. Nucleic Acids Res. 1995, 23, 459–463. [Google Scholar] [CrossRef]
- Shi, W.; Inoue, M.; Minami, M.; Takeda, K.; Matsumoto, M.; Matsuda, Y.; Matsuda, Y.; Kishimoto, T.; Akira, S. The genomic structure and chromosomal lozalization of the mouse STAT3 gene. Int. Immunol. 1996, 8, 1205–1211. [Google Scholar] [CrossRef]
- Sugiyama, T.; Nishio, Y.; Kishimoto, T.; Akira, S. Identification of alternative splicing form of Stat2. FEBS Lett. 1996, 381, 191–194. [Google Scholar] [CrossRef]
- Schaefer, T.S.; Sanders, L.K.; Nathans, D. Cooperative transcriptional activity of Jun and Stat3 beta, a short form of Stat3. Proc. Natl. Acad. Sci. USA 1995, 92, 9097–9101. [Google Scholar] [CrossRef] [PubMed]
- Schindler, C.; Fu, X.Y.; Improta, T.; Aebersold, R.; Darnell, J.E., Jr. Proteins of transcription factor ISGF-3: One gene encodes the 91-and 84-kDa ISGF-3 proteins that are activated by interferon alpha. Proc. Natl. Acad. Sci. USA 1992, 89, 7836–7839. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Z. STAT1 in cancer: Friend or foe? Discov. Med. 2017, 24, 19–29. [Google Scholar]
- Dimberg, A.; Karlberg, I.; Nilsson, K.; Öberg, F. Ser727/Tyr701-phosphorylated Stat1 is required for the regulation of c-Myc, cyclins, and p27Kip1 associated with ATRA-induced G0/G1 arrest of U-937 cells. Blood 2003, 102, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Schlee, M.; Hölzel, M.; Bernard, S.; Mailhammer, R.; Schuhmacher, M.; Reschke, J.; Eick, D.; Marinkovic, D.; Wirth, T.; Rosenwald, A.; et al. C-myc activation impairs the NF-kappaB and the interferon response: Implications for the pathogenesis of Burkitt’s lymphoma. Int. J. Cancer 2007, 120, 1387–1395. [Google Scholar] [CrossRef]
- Dimberg, A.; Nilsson, K.; Öberg, F. Phosphorylation-deficient Stat1 inhibits retinoic acid-induced differentiation and cell cycle arrest in U-937 monoblasts. Blood 2000, 96, 2870–2878. [Google Scholar] [CrossRef] [PubMed]
- Takayanagi, H.; Kim, S.; Koga, T.; Taniguchi, T. Stat1-mediated cytoplasmic attenuation in osteoimmunology. J. Cell. Biochem. 2005, 94, 232–240. [Google Scholar] [CrossRef]
- Lee, C.K.; Smith, E.; Gimeno, R.; Gertner, R.; Levy, D.E. STAT1 affects lymphocyte survival and proliferation partially independent of its role downstream of IFN-γ. J. Immunol. 2000, 164, 1286–1292. [Google Scholar] [CrossRef]
- Wold, W.S.; Toth, K. Adenovirus vectors for gene therapy, vaccination and cancer gene therapy. Curr. Gene Ther. 2013, 13, 421–433. [Google Scholar] [CrossRef]
- Gu, W.; Roeder, R.G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef]
- Dovhey, S.E.; Ghosh, N.S.; Wright, K.L. Loss of interferon-gamma inducibility of TAP1 and LMP2 in a renal cell carcinoma cell line. Cancer Res. 2000, 60, 5789–5796. [Google Scholar] [PubMed]
- Chatterjee-Kishore, M.; Kishore, R.; Hicklin, D.J.; Marincola, F.M.; Ferrone, S. Different Requirements for Signal Transducer and Activator of Transcription 1α and Interferon Regulatory Factor 1 in the Regulation of Low Molecular Mass Polypeptide 2 and Transporter Associated with Antigen Processing 1 Gene Expression. J. Biol. Chem. 1998, 273, 16177–16183. [Google Scholar] [CrossRef]
- Ding, Y.; Yang, M.; She, S.; Min, H.; Xv, X.; Ran, X.; Wu, Y.; Wang, W.; Wang, L.; Yi, L.; et al. ITRAQ-based quantitative proteomic analysis of cervical cancer. Int. J. Oncol. 2015, 46, 1748–1758. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, T.; Sabitha, K.; Vijayalakshmi, N.; Shirley, S.; Bose, M.V.; Gopal, G.; Selvaluxmy, G. Identification and validation of genes involved in cervical tumourigenesis. BMC Cancer 2011, 11, 80. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Fang, Y.; Wu, K.; Liu, Y.; Zhang, W. Comprehensive gene and pathway analysis of cervical cancer progression. Oncol. Lett. 2020, 19, 3316–3333. [Google Scholar] [CrossRef]
- Buttarelli, M.; Babini, G.; Raspaglio, G.; Filippetti, F.; Battaglia, A.; Ciucci, A.; Ferrandina, G.; Petrillo, M.; Marino, C.; Mancuso, M.; et al. A combined ANXA2-NDRG1-STAT1 gene signature predicts response to chemoradiotherapy in cervical cancer. J. Exp. Clin. Cancer Res. 2019, 38, 279. [Google Scholar] [CrossRef]
- Okunade, K.S. Human papillomavirus and cervical cancer. J. Obstet. Gynaecol. 2020, 40, 602–608. [Google Scholar] [CrossRef]
- Chang, Y.E.; Laimins, L.A. Microarray Analysis Identifies Interferon-Inducible Genes and Stat-1 as Major Transcriptional Targets of Human Papillomavirus Type 31. J. Virol. 2000, 74, 4174–4182. [Google Scholar] [CrossRef]
- Hong, S.; Mehta, K.P.; Laimins, L.A. Suppression of STAT-1 Expression by Human Papillomaviruses Is Necessary for Differentiation-Dependent Genome Amplification and Plasmid Maintenance. J. Virol. 2011, 85, 9486–9494. [Google Scholar] [CrossRef]
- Li, S.; Labrecque, S.; Gauzzi, M.C.; Cuddihy, A.R.; Wong, A.H.T.; Pellegrini, S.; Matlashewski, G.J.; Koromilas, A.E. The human papilloma virus (HPV)-18 E6 oncoprotein physically associates with Tyk2 and impairs Jak-STAT activation by interferon-α. Oncogene 1999, 18, 5727–5737. [Google Scholar] [CrossRef]
- Nees, M.; Geoghegan, J.M.; Hyman, T.; Frank, S.; Miller, L.; Woodworth, C.D. Papillomavirus Type 16 Oncogenes Downregulate Expression of Interferon-Responsive Genes and Upregulate Proliferation-Associated and NF-κB-Responsive Genes in Cervical Keratinocytes. J. Virol. 2001, 75, 4283–4296. [Google Scholar] [CrossRef] [PubMed]
- Schindler, C.; Plumlee, C. Inteferons pen the JAK–STAT pathway. Semin. Cell Dev. Biol. 2008, 19, 311–318. [Google Scholar]
- Wang, Y.; Song, Q.; Huang, W.; Lin, Y.; Wang, X.; Wang, C.; Willard, B.; Zhao, C.; Nan, J.; Holvey-Bates, E.; et al. A virus-induced conformational switch of STAT1-STAT2 dimers boosts antiviral defenses. Cell Res. 2021, 31, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Li, S.; Cha, E.; Schindler, C. Immune Response in Stat2 Knockout Mice. Immunity 2000, 13, 795–804. [Google Scholar] [CrossRef]
- Liang, Z.; Gao, L.H.; Cao, L.J.; Feng, D.Y.; Cao, Y.; Luo, Q.Z.; Yu, P.; Li, M. Detection of STAT2 in early stage of cervical premalignancy and in cervical cancer. Asian Pac. J. Trop. Med. 2012, 5, 738–742. [Google Scholar] [CrossRef]
- Antonsson, A.; Payne, E.; Hengst, K.; McMillan, D.N.A.J. The Human Papillomavirus Type 16 E7 Protein Binds Human Interferon Regulatory Factor-9 via a Novel PEST Domain Required for Transformation. J. Interf. Cytokine Res. 2006, 26, 455–461. [Google Scholar] [CrossRef]
- Maritano, D.; Sugrue, M.L.; Tininini, S.; Dewilde, S.; Strobl, B.; Fu, X.; Murray-Tait, V.; Chiarle, R.; Poli, V. The STAT3 isoforms alpha and beta have unique and specific functions. Nat. Immunol. 2004, 5, 401–409. [Google Scholar] [CrossRef]
- Caldenhoven, E.; van Dijk, T.B.; Solari, R.; Armstrong, J.; Raaijmakers, J.A.; Lammers, J.W.J.; Koenderman, L.; de Groot, R.P. STAT3beta, a splice variant of transcription factor STAT3, is a dominant negative regulator of transcription. J. Biol. Chem. 1996, 271, 13221–13227. [Google Scholar] [CrossRef]
- Schaefer, T.S.; Sanders, L.K.; Park, O.K.; Nathans, D. Functional differences between Stat3alpha and Stat3beta. Mol. Cell Biol. 1997, 17, 53077–59101. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Behrmann, I.; Müller-Newen, G.; Schaper, F.; Graeve, L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J. 1998, 334 Pt 2, 297–314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhou, L.; Xu, Y.; Yang, M.; Xu, Y.; Komaniecki, G.P.; Kosciuk, T.; Chen, X.; Lu, X.; Zou, X.; et al. A STAT3 palmitoylation cycle promotes TH17 differentiation and colitis. Nature 2020, 586, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Kortylewski, M.; Kujawski, M.; Wang, T.; Wei, S.; Zhang, S.; Pilon-Thomas, S.; Niu, G.; Kay, H.; Mulé, J.; Kerr, W.; et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat. Med. 2005, 11, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-Derived Suppressor Cells: Linking Inflammation and Cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Yu, H.; Kortylewski, M.; Pardoll, D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51. [Google Scholar] [CrossRef]
- Catlett-Falcone, R.; Landowski, T.H.; Oshiro, M.M.; Turkson, J.; Levitzki, A.; Savino, R.; Ciliberto, G.; Moscinski, L.; Fernández-Luna, J.L.; Nuñez, G.; et al. Constitutive Activation of Stat3 Signaling Confers Resistance to Apoptosis in Human U266 Myeloma Cells. Immunity 1999, 10, 105–115. [Google Scholar] [CrossRef]
- Avalle, L.; Camporeale, A.; Camperi, A.; Poli, V. STAT3 in cancer: A double edged sword. Cytokine 2017, 98, 42–50. [Google Scholar] [CrossRef]
- Niu, G.; Heller, R.; Catlett-Falcone, R.; Coppola, D.; Jaroszeski, M.; Dalton, W.; Jove, R.; Yu, H. Gene therapy with dominant-negative Stat3 suppresses growth of the murine melanoma B16 tumor in vivo. Cancer Res. 1999, 59, 5059–5063. [Google Scholar]
- Lee, J.-L.; Wang, M.-J.; Chen, J.-Y. Acetylation and activation of STAT3 mediated by nuclear translocation of CD44. J. Cell Biol. 2009, 185, 949–957. [Google Scholar] [CrossRef]
- Wu, L.; Shen, B.; Li, J.; Zhang, H.; Zhang, K.; Yang, Y.; Zu, Z.; Shen, D.; Luo, M. STAT3 exerts pro-tumor and anti-autophagy roles in cervical cancer. Diagn. Pathol. 2022, 17, 13. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Mahata, S.; Shishodia, G.; Pandey, A.; Tyagi, A.; Vishnoi, K.; Basir, S.F.; Das, B.C.; Bharti, A.C. Functional Regulatory Role of STAT3 in HPV16-Mediated Cervical Carcinogenesis. PLoS ONE 2013, 8, e67849. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Shishodia, G.; Mahata, S.; Hedau, S.; Pandey, A.; Bhambhani, S.; Batra, S.; Basir, S.F.; Das, B.C.; Bharti, A.C. Aberrant expression and constitutive activation of STAT3 in cervical carcinogenesis: Implications in high-risk human papillomavirus infection. Mol. Cancer 2010, 9, 282. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-L.; Hsieh, F.-C.; Lieblein, J.C.; Brown, J.; Chan, C.; Wallace, J.A.; Cheng, G.; Hall, B.M.; Lin, J. Stat3 activation in human endometrial and cervical cancers. Br. J. Cancer 2007, 96, 591–599. [Google Scholar] [CrossRef]
- Shi, S.; Ma, H.Y.; Zhang, Z.G. Clinicopathological and prognostic value of STAT3/p-STAT3 in cervical cancer: A meta and bioinformatics analysis. Pathol. Res. Pract. 2021, 227, 153624. [Google Scholar] [CrossRef] [PubMed]
- de Arellano, A.R.; Lopez-Pulido, E.I.; Martínez-Neri, P.A.; Chávez, C.E.; Lucano, R.G.; Fafutis-Morris, M.; Aguilar-Lemarroy, A.; Muñoz-Valle, J.F.; Pereira-Suárez, A.L. STAT3 activation is required for the antiapoptotic effects of prolactin in cervical cancer cells. Cancer Cell Int. 2015, 15, 83. [Google Scholar] [CrossRef]
- Morgan, E.L.; Macdonald, A. Autocrine STAT3 activation in HPV positive cervical cancer through a virus-driven Rac1—NFκB—IL-6 signalling axis. PLoS Pathog. 2019, 15, e1007835. [Google Scholar] [CrossRef]
- Shukla, S.; Jadli, M.; Thakur, K.; Shishodia, G.; Mahata, S.; Basir, S.F.; Das, B.C.; Bharti, A.C. Level of phospho-STAT3 (Tyr705) correlates with copy number and physical state of human papillomavirus 16 genome in cervical precancer and cancer lesions. PLoS ONE 2019, 14, e0222089. [Google Scholar] [CrossRef]
- Morgan, E.; Wasson, C.W.; Hanson, L.; Kealy, D.; Pentland, I.; McGuire, V.; Scarpini, C.; Coleman, N.; Arthur, J.S.C.; Parish, J.L.; et al. STAT3 activation by E6 is essential for the differentiation-dependent HPV18 life cycle. PLoS Pathog. 2018, 14, e1006975. [Google Scholar] [CrossRef]
- Fan, Z.; Cui, H.; Xu, X.; Lin, Z.; Zhang, X.; Kang, L.; Han, B.; Meng, J.; Yan, Z.; Yan, X.; et al. MiR-125a suppresses tumor growth, invasion and metastasis in cervical cancer by targeting STAT3. Oncotarget 2015, 6, 25266–25280. [Google Scholar] [CrossRef]
- Kim, L.; Park, S.A.; Park, H.; Kim, H.; Heo, T.H. Bazedoxifene, a GP130 inhibitor, modulates EMT signaling and exhibits antitumor effects in HPV-positive cervical cancer. Int. J. Mol. Sci. 2021, 22, 8693. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.-L.; Rao, W. SiRNA interfering STAT3 enhances DDP sensitivity in cervical cancer cells. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 4098–4106. [Google Scholar] [PubMed]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting Cancer Stem Cell Pathways for Cancer Therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Miyagi, T.; Gil, M.P.; Wang, X.; Louten, J.; Chu, W.-M.; Biron, C.A. High basal STAT4 balanced by STAT1 induction to control type 1 interferon effects in natural killer cells. J. Exp. Med. 2007, 204, 2383–2396. [Google Scholar] [CrossRef]
- Thieu, V.T.; Yu, Q.; Chang, H.-C.; Yeh, N.; Nguyen, E.T.; Sehra, S.; Kaplan, M.H. Signal Transducer and Activator of Transcription 4 Is Required for the Transcription Factor T-bet to Promote T Helper 1 Cell-Fate Determination. Immunity 2008, 29, 679–690. [Google Scholar] [CrossRef]
- Luo, J.; Huang, Q.; Lin, X.; Wei, K.; Ling, Y.; Su, S.; Cao, Y.; Luo, J.; Pan, D.; Dang, Y.; et al. STAT4 expression is correlated with clinicopathological characteristics of cervical lesions. Int. J. Clin. Exp. Pathol. 2016, 9, 3751–3758. [Google Scholar]
- Ibarra Sierra, E.; Díaz Chávez, J.; Cortés-Malagón, E.M.; Uribe-Figueroa, L.; Hidalgo-Miranda, A.; Lambert, P.F.; Gariglio, P. Differential gene expression between skin and cervix induced by the E7 oncoprotein in a transgenic mouse model. Virology 2012, 433, 337–345. [Google Scholar] [CrossRef]
- Grimley, P.M.; Dong, F.; Rui, H. Stat5a and Stat5b: Fraternal twins of signal transduction and transcriptional activation. Cytokine Growth Factor Rev. 1999, 10, 131–157. [Google Scholar] [CrossRef]
- Schindler, C.; Levy, D.E.; Decker, T. JAK-STAT Signaling: From Interferons to Cytokines. J. Biol. Chem. 2007, 282, 20059–20063. [Google Scholar] [CrossRef]
- Lin, J.-X.; Du, N.; Li, P.; Kazemian, M.; Gebregiorgis, T.; Spolski, R.; Leonard, W.J. Critical functions for STAT5 tetramers in the maturation and survival of natural killer cells. Nat. Commun. 2017, 8, 1320. [Google Scholar] [CrossRef]
- Azam, M.; Erdjument-Bromage, H.; Kreider, B.; Xia, M.; Quelle, F.; Basu, R.; Saris, C.; Tempst, P.; Ihle, J.; Schindler, C. Interleukin-3 signals through multiple isoforms of Stat5. EMBO J. 1995, 14, 1402–1411. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Robinson, G.W.; Wagner, K.U.; Garrett, L.; Wynshaw-Boris, A.; Hennighausen, L. Stat5a is mandatory for adult mammary gland development and lactogenesis. Genes Dev. 1997, 11, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.X.; Leonard, W.J. The role of Stat5a and Stat5b in signaling by IL-2 family cytokines. Oncogene 2000, 19, 2566–2576. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-X.; Li, P.; Liu, D.; Jin, H.T.; He, J.; Rasheed, M.A.U.; Rochman, Y.; Wang, L.; Cui, K.; Liu, C.; et al. Critical Role of STAT5 Transcription Factor Tetramerization for Cytokine Responses and Normal Immune Function. Immunity 2012, 36, 586–599. [Google Scholar] [CrossRef] [PubMed]
- Pericle, F.; Kirken, R.A.; Bronte, V.; Sconocchia, G.; DaSilva, L.; Segal, D.M. Immunocompromised tumor-bearing mice show a selective loss of STAT5a/b expression in T and B lymphocytes. J. Immunol. 1997, 159, 2580–2585. [Google Scholar] [CrossRef]
- Refaeli, Y.; Van Parijs, L.; London, C.A.; Tschopp, J.; Abbas, A.K. Biochemical Mechanisms of IL-2–Regulated Fas-Mediated T Cell Apoptosis. Immunity 1998, 8, 615–623. [Google Scholar] [CrossRef]
- Morgan, E.L.; Macdonald, A. JAK2 Inhibition Impairs Proliferation and Sensitises Cervical Cancer Cells to Cisplatin-Induced Cell Death. Cancers 2019, 11, 1934. [Google Scholar] [CrossRef]
- Sobti, R.C.; Singh, N.; Hussain, S.; Suri, V.; Bharadwaj, M.; Das, B.C. Deregulation of STAT-5 isoforms in the development of HPV-mediated cervical carcinogenesis. J. Recept. Signal Transduct. 2010, 30, 178–188. [Google Scholar] [CrossRef]
- Hong, S.; Laimins, L.A. The JAK-STAT transcriptional regulator, STAT-5, activates the ATM DNA damage pathway to induce HPV 31 genome amplification upon epithelial differentiation. PLoS Pathog. 2013, 9, e1003295. [Google Scholar] [CrossRef]
- Quelle, F.W.; Shimoda, K.; Thierfelder, W.; Fischer, C.; Kim, A.; Ruben, S.M.; Cleveland, J.L.; Pierce, J.H.; Keegan, A.D.; Nelms, K.; et al. Cloning of Murine Stat6 and Human Stat6, Stat Proteins That Are Tyrosine Phosphorylated in Responses to IL-4 and IL-3 but Are Not Required for Mitogenesis. Mol. Cell. Biol. 1995, 15, 3336–3343. [Google Scholar] [CrossRef]
- Mikita, T.; Daniel, C.; Wu, P.; Schindler, U. Mutational Analysis of the STAT6 SH2 Domain. J. Biol. Chem. 1998, 273, 17634–17642. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Sun, H.; You, F.; Sun, W.; Zhou, X.; Chen, L.; Yang, J.; Wang, Y.; Tang, H.; Guan, Y.; et al. Activation of STAT6 by STING Is Critical for Antiviral Innate Immunity. Cell 2011, 147, 436–446. [Google Scholar] [CrossRef]
- Duetsch, G.; Illig, T.; Loesgen, S.; Rohde, K.; Klopp, N.; Herbon, N.; Gohlke, H.; Altmueller, J.; Wjst, M. STAT6 as an asthma candidate gene: Polymorphism-screening, association and haplotype analysis in a Caucasian sib-pair study. Hum. Mol. Genet. 2002, 11, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Paul, W.E. CD4 T cells: Fates, functions, and faults. Blood 2008, 112, 1557–1569. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, K.; van Deursent, J.; Sangster, M.Y.; Sarawar, S.R.; Carson, R.T.; Tripp, R.A.; Chu, C.; Quelle, F.W.; Nosaka, T.; Vignali, D.A.A.; et al. Lack of IL-4-induced Th2 response and IgE class switching in mice with disrupted Stat6 gene. Nature 1996, 380, 630–633. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, M.H.; Schindler, U.; Smiley, S.T.; Grusby, M.J. Stat6 Is Required for Mediating Responses to IL-4 and for the Development of Th2 Cells. Immunity 1996, 4, 313–319. [Google Scholar] [CrossRef]
- Zhang, W.; Wu, X.; Hu, L.; Ma, Y.; Xiu, Z.; Huang, B.; Feng, Y.; Tang, X. Overexpression of human papillomavirus type 16 oncoproteins enhances epithelial-mesenchymal transition via STAT3 signaling pathway in non-small cell lung cancer cells. Oncol. Res. 2017, 25, 843–852. [Google Scholar] [CrossRef]
- Li, Z.; Guan, Y.Q.; Liu, J.M. The role of STAT-6 as a key transcription regulator in HeLa cell death induced by IFN-γ/TNF-α co-immobilized on nanoparticles. Biomaterials 2014, 35, 5016–5027. [Google Scholar] [CrossRef]
- Yoshimura, A.; Ohkubo, T.; Kiguchi, T.; Jenkins, N.A.; Gilbert, D.J.; Copeland, N.G.; Hara, T.; Miyajima, A. A novel cytokine-inducible gene CIS encodes an SH2-containing protein that binds to tyrosine-phosphorylated interleukin 3 and erythropoietin receptors. EMBO J. 1995, 14, 2816–2826. [Google Scholar] [CrossRef]
- Kershaw, N.; Murphy, J.; Liau, N.; Varghese, L.N.; Laktyushin, A.; Whitlock, E.L.; Lucet, I.S.; Nicola, N.A.; Babon, J.J. SOCS3 binds specific receptor–JAK complexes to control cytokine signaling by direct kinase inhibition. Nat. Struct. Mol. Biol. 2013, 20, 469–476. [Google Scholar] [CrossRef]
- Yasukawa, H.; Misawa, H.; Sakamoto, H.; Masuhara, M.; Sasaki, A.; Wakioka, T.; Ohtsuka, S.; Imaizumi, T.; Matsuda, T.; Ihle, J.N.; et al. The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop. EMBO J. 1999, 18, 1309–1320. [Google Scholar] [CrossRef] [PubMed]
- Tamiya, T.; Kashiwagi, I.; Takahashi, R.; Yasukawa, H.; Yoshimura, A. Suppressors of cytokine signaling (SOCS) proteins and JAK/STAT pathways: Regulation of T-cell inflammation by SOCS1 and SOCS3. Arter. Thromb. Vasc. Biol. 2011, 31, 980–985. [Google Scholar] [CrossRef] [PubMed]
- Okumura, F.; Joo-Okumura, A.; Nakatsukasa, K.; Kamura, T. The role of cullin 5-containing ubiquitin ligases. Cell Div. 2016, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Sobti, R.C.; Singh, N.; Hussain, S.; Suri, V.; Nijhawan, R.; Bharti, A.C.; Bharadwaj, M.; Das, B.C. Aberrant promoter methylation and loss of Suppressor of Cytokine Signalling-1 gene expression in the development of uterine cervical carcinogenesis. Cell. Oncol. 2011, 34, 533–543. [Google Scholar] [CrossRef]
- Kim, M.H.; Kim, M.S.; Kim, W.; Kang, M.A.; Cacalano, N.A.; Kang, S.B.; Shin, Y.J.; Jeong, J.H. Suppressor of cytokine signaling (SOCS) genes are silenced by DNA hypermethylation and histone deacetylation and regulate response to radiotherapy in cervical cancer cells. PLoS ONE 2015, 10, e0123133. [Google Scholar] [CrossRef]
- Kamio, M.; Yoshida, T.; Ogata, H.; Douchi, T.; Nagata, Y.; Inoue, M.; Hasegawa, M.; Yonemitsu, Y.; Yoshimura, A. SOC1 inhibits HPV-E7-mediated transformation by inducing degradation of E7 protein. Oncogene 2004, 23, 3107–3115. [Google Scholar] [CrossRef]
- Mohr, S.E.; Boswell, R.E. Zimp encodes a homologue of mouse Miz1 and PIAS3 and is an essential gene in Drosophila melanogaster. Gene 1999, 229, 109–116. [Google Scholar] [CrossRef]
- Wu, H.; Liu, X.; Jaenisch, R.; Lodish, H.F. Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor. Cell 1995, 83, 59–67. [Google Scholar] [CrossRef]
- Sonnenblick, A.; Levy, C.; Razin, E. Interplay between MITF, PIAS3, and STAT3 in mast cells and melanocytes. Mol. Cell. Biol. 2004, 24, 10584–10592. [Google Scholar] [CrossRef][Green Version]
- Rogers, R.S.; Horvath, C.M.; Matunis, M.J. SUMO Modification of STAT1 and Its Role in PIAS-mediated Inhibition of Gene Activation. J. Biol. Chem. 2003, 278, 30091–30097. [Google Scholar] [CrossRef]
- Tussié-Luna, M.I.; Bayarsaihan, D.; Seto, E.; Ruddle, F.H.; Roy, A.L. Physical and functional interactions of histone deacetylase 3 with TFII-I family proteins and PIASxbeta. Proc. Natl. Acad. Sci. USA 2002, 99, 12807–12812. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, S.; Bruhn, L.; Sieber, H.; Pichler, A.; Melchior, F.; Grosschedl, R. PIASy, a nuclear matrix–associated SUMO E3 ligase, represses LEF1 activity by sequestration into nuclear bodies. Genes Dev. 2001, 15, 3088–3103. [Google Scholar] [CrossRef] [PubMed]
- Hoeve, J.T.; Ibarra-Sanchez, M.d.J.; Fu, Y.; Zhu, W.; Tremblay, M.; David, M.; Shuai, K. Identification of a Nuclear Stat1 Protein Tyrosine Phosphatase. Mol. Cell. Biol. 2002, 22, 5662–5668. [Google Scholar] [CrossRef] [PubMed]
- Ram, P.A.; Waxman, D.J. Interaction of growth hormone-activated STATs with SH2-containing phosphotyrosine phosphatase SHP-1 and nuclear JAK2 tyrosine kinase. J. Biol. Chem. 1997, 272, 17694–17702. [Google Scholar] [CrossRef] [PubMed]
- Irie-Sasaki, J.; Sasaki, T.; Matsumoto, W.; Opavsky, A.; Cheng, M.; Welstead, G.; Griffiths, E.; Krawczyk, C.; Richardson, C.D.; Aitken, K.; et al. CD45 is a JAK phosphatase and negatively regulates cytokine receptor signalling. Nature 2001, 409, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.P.; Andersen, J.N.; Cheng, A.; Tremblay, M.L.; Horvath, C.M.; Parisien, J.-P.; Salmeen, A.; Barford, D.; Tonks, N.K. TYK2 and JAK2 Are Substrates of Protein-tyrosine Phosphatase 1B. J. Biol. Chem. 2001, 276, 47771–47774. [Google Scholar] [CrossRef]
- Klingmüller, U.; Lorenz, U.; Cantley, L.C.; Neel, B.G.; Lodish, H.F. Specific recruitment of SH-PTP1 to the erythropoietin receptor causes inactivation of JAK2 and termination of proliferative signals. Cell 1995, 80, 729–738. [Google Scholar] [CrossRef]
- You, M.; Yu, D.-H.; Feng, G.-S. Shp-2 Tyrosine Phosphatase Functions as a Negative Regulator of the Interferon-Stimulated Jak/STAT Pathway. Mol. Cell. Biol. 1999, 19, 2416–2424. [Google Scholar] [CrossRef]
- Iuliano, M.; Mangino, G.; Chiantore, M.V.; Di Bonito, P.; Rosa, P.; Affabris, E.; Romeo, G. Virus-Induced Tumorigenesis and IFN System. Biology 2021, 10, 994. [Google Scholar] [CrossRef]
- Gupta, S.; Kumar, P.; Das, B.C. HPV: Molecular pathways and targets. Curr. Probl. Cancer 2018, 42, 161–174. [Google Scholar] [CrossRef]
- Saha, A.; Kaul, R.; Murakami, M.; Robertson, E.S. Tumor viruses and cancer biology: Modulating signaling pathways for therapeutic intervention. Cancer Biol. Ther. 2010, 10, 961–978. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chen, Z.J. The cGAS–cGAMP–STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA sensing by the cGAS–STING pathway in health and disease. Nat. Rev. Genet. 2019, 20, 657–674. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.; Gray, E.E.; Brunette, R.L.; Stetson, D.B. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 2015, 350, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, M.H.; Bortnik, V.; McMillan, N.A.; Idris, A. cGAS-STING responses are dampened in high-risk HPV type 16 positive head and neck squamous cell carcinoma cells. Microb. Pathog. 2019, 132, 162–165. [Google Scholar] [CrossRef]
- Scott, M.L.; Woodby, B.L.; Ulicny, J.; Raikhy, G.; Orr, A.W.; Songock, W.K.; Bodily, J.M. Human Papillomavirus 16 E5 Inhibits Interferon Signaling and Supports Episomal Viral Maintenance. J. Virol. 2020, 94, e01582-19. [Google Scholar] [CrossRef] [PubMed]
- Reiser, J.; Hurst, J.; Voges, M.; Krauss, P.; Münch, P.; Iftner, T.; Stubenrauch, F. High-Risk Human Papillomaviruses Repress Constitutive Kappa Interferon Transcription via E6 To Prevent Pathogen Recognition Receptor and Antiviral-Gene Expression. J. Virol. 2011, 85, 11372–11380. [Google Scholar] [CrossRef]
- Sunthamala, N.; Thierry, F.; Teissier, S.; Pientong, C.; Kongyingyoes, B.; Tangsiriwatthana, T.; Sangkomkamhang, U.; Ekalaksananan, T. E2 Proteins of High Risk Human Papillomaviruses Down-Modulate STING and IFN-κ Transcription in Keratinocytes. PLoS ONE 2014, 9, e91473. [Google Scholar] [CrossRef]
- Ronco, L.V.; Karpova, A.Y.; Vidal, M.; Howley, P.M. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev. 1998, 12, 2061–2072. [Google Scholar] [CrossRef]
- Park, J.S.; Kim, E.J.; Kwon, H.J.; Hwang, E.S.; Namkoong, S.E.; Um, S.J. Inactivation of interferon regulatory factor-1 tumor suppressor protein by HPV E7 oncoprotein. Implication for the E7-mediated immune evasion mechanism in cervical carcinogenesis. J. Biol. Chem. 2000, 275, 6764–6769. [Google Scholar] [CrossRef]
- Perea, S.E.; Massimi, P.; Banks, L. Human papillomavirus type 16 E7 impairs the activation of the interferon regulatory factor-1. Int. J. Mol. Med. 2000, 5, 661–666. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Lv, X.; Huang, C.; Angeletti, P.C.; Hua, G.; Dong, J.; Zhou, J.; Wang, Z.; Ma, B.; Chen, X.; et al. A Human Papillomavirus-Independent Cervical Cancer Animal Model Reveals Unconventional Mechanisms of Cervical Carcinogenesis. Cell Rep. 2019, 26, 2636–2650.e5. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.Y.; Cao, C.; Liu, L. Interferon α induces the apoptosis of cervical cancer HeLa cells by activating both the intrinsic mitochondrial pathway and endoplasmic reticulum stress-induced pathway. Int. J. Mol. Sci. 2016, 17, 1832. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.L.; Tan, H.X.; Niu, T.T.; Liu, Y.K.; Gu, C.J.; Li, D.J.; Li, M.-Q.; Wang, H.-Y. The IFN-γ-IDO1-kynureine pathway-induced autophagy in cervical cancer cell promotes phagocytosis of macrophage. Int. J. Biol. Sci. 2021, 17, 339. [Google Scholar] [CrossRef]
- Cannella, F.; Scagnolari, C.; Selvaggi, C.; Stentella, P.; Recine, N.; Antonelli, G.; Pierangeli, A. Interferon lambda 1 expression in cervical cells differs between low-risk and high-risk human papillomavirus-positive women. Med. Microbiol. Immunol. 2014, 203, 177–184. [Google Scholar] [CrossRef]
- Bhattacharjee, R.; Das, S.S.; Biswal, S.S.; Nath, A.; Das, D.; Basu, A.; Malik, S.; Kumar, L.; Kar, S.; Singh, S.K.; et al. Mechanistic role of HPV-associated early proteins in cervical cancer: Molecular pathways and targeted therapeutic strategies. Crit. Rev. Oncol. 2022, 174, 103675. [Google Scholar] [CrossRef]
- Lichtig, H.; Gilboa, D.A.; Jackman, A.; Gonen, P.; Levav-Cohen, Y.; Haupt, Y.; Sherman, L. HPV16 E6 augments Wnt signaling in an E6AP-dependent manner. Virology 2010, 396, 47–58. [Google Scholar] [CrossRef]
- Drews, C.M.; Case, S.; Pol, S.B.V. E6 proteins from high-risk HPV, low-risk HPV, and animal papillomaviruses activate the Wnt/β-catenin pathway through E6AP-dependent degradation of NHERF1. PLoS Pathog. 2019, 15, e1007575. [Google Scholar] [CrossRef]
- Barker, N.; Clevers, H. Catenins, Wnt signaling and cancer. Bioessays 2000, 22, 961–965. [Google Scholar] [CrossRef]
- Manzo-Merino, J.; Contreras-Paredes, A.; Vázquez-Ulloa, E.; Rocha-Zavaleta, L.; Fuentes-Gonzalez, A.M.; Lizano, M. The Role of Signaling Pathways in Cervical Cancer and Molecular Therapeutic Targets. Arch. Med. Res. 2014, 45, 525–539. [Google Scholar] [CrossRef]
- Ramachandran, I.; Thavathiru, E.; Ramalingam, S.; Natarajan, G.; Mills, W.K.; Benbrook, D.M.; Zuna, R.; Lightfoot, S.; Reis, A.; Anant, S.; et al. Wnt inhibitory factor 1 induces apoptosis and inhibits cervical cancer growth, invasion and angiogenesis in vivo. Oncogene 2011, 31, 2725–2737. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Takaoka, A.S.; Naishiro, Y.; Hayashi, R.; Maruyama, K.; Maesawa, C.; Ochiai, A.; Hirohashi, S. Transactivation of the multidrug resistance 1 gene by T-cell factor 4/beta-catenin complex in early colorectal carcinogenesis. Cancer Res. 2000, 60, 4761–4766. [Google Scholar]
- Yao, R.; Cooper, G.M. Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science 1995, 267, 2003–2006. [Google Scholar] [CrossRef] [PubMed]
- Kauffmann-Zeh, A.; Rodriguez-Viciana, P.; Ulrich, E.; Gilbert, C.; Coffer, P.; Downward, J.; Evan, G. Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature 1997, 385, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.M.; Fuhrman, C.B.; Planelles, V.; Peltier, M.R.; Gaffney, D.K.; Soisson, A.P.; Dodson, M.K.; Tolley, H.D.; Green, C.L.; Zempolich, K.A. Phosphatidylinositol 3-Kinase Inhibition by LY294002 Radiosensitizes Human Cervical Cancer Cell Lines. Clin. Cancer Res. 2006, 12, 250–256. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L.; Akagi, K.; Xiao, W.; Jiang, B.; Pickard, R.K.; Li, J.; Swanson, B.J.; Agrawal, A.D.; Zucker, M.; Stache-Crain, B.; et al. Human papillomavirus and the landscape of secondary genetic alterations in oral cancers. Genome Res. 2018, 29, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Menges, C.W.; Baglia, L.A.; Lapoint, R.; McCance, D.J. Human Papillomavirus Type 16 E7 Up-regulates AKT Activity through the Retinoblastoma Protein. Cancer Res. 2006, 66, 5555–5559. [Google Scholar] [CrossRef]
- Pim, D.; Massimi, P.; Dilworth, S.M.; Banks, L. Activation of the protein kinase B pathway by the HPV-16 E7 oncoprotein occurs through a mechanism involving interaction with PP2A. Oncogene 2005, 24, 7830–7838. [Google Scholar] [CrossRef]
- Lu, Z.; Hu, X.; Li, Y.; Zheng, L.; Zhou, Y.; Jiang, H.; Ning, T.; Basang, Z.; Zhang, C.; Ke, Y. Human Papillomavirus 16 E6 oncoprotein interferences with insulin signaling pathway by binding to tuberin. J. Biol. Chem. 2004, 276, 35664–35670. [Google Scholar] [CrossRef]
- Contreras-Paredes, A.; De la Cruz-Hernández, E.; Martínez-Ramírez, I.; Dueñas-González, A.; Lizano, M. E6 variants of human papillomavirus 18 differentially modulate the protein kinase B/phosphatidylinositol 3-kinase (akt/PI3K) signaling pathway. Virology 2009, 383, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Kohno, M.; Pouyssegur, J. Targeting the ERK signaling pathway in cancer therapy. Ann. Med. 2006, 38, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Pagès, G.; Lenormand, P.; L’Allemain, G.; Chambard, J.C.; Meloche, S.; Pouysségur, J. Mitogen-activated protein kinases p42mapk and p44mapk are required for fibroblast proliferation. Proc. Natl. Acad. Sci. USA 1993, 90, 8319–8323. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Liu, W.-Z.; Liu, T.; Feng, X.; Yang, N.; Zhou, H.-F. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept. Signal Transduct. 2015, 35, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Lewis, T.S.; Shapiro, P.S.; Ahn, N.G. Signal Transduction through MAP Kinase Cascades. Adv. Cancer Res. 1998, 74, 49–139. [Google Scholar] [PubMed]
- Branca, M.; Ciotti, M.; Santini, D.; Bonito, L.D.; Benedetto, A.; Giorgi, C.; Paba, P.; Favalli, C.; Costa, S.; Agarossi, A.; et al. Activation of the ERK/MAP kinase pathway in cervical intraepithelial neoplasia is related to grade of the lesion but not to high-risk human papillomavirus, virus clearance, or prognosis in cervical cancer. Am. J. Clin. Pathol. 2004, 122, 902–911. [Google Scholar] [CrossRef]
- Fanger, G.R. Regulation of the MAPK family members: Role of subcellular localization and architectural organization. Histol. Histopathol. 1999, 14, 887–894. [Google Scholar]
- Kyriakis, J.M. Activation of the AP-1 transcription factor by inflammatory cytokines of the TNF family. Gene Expr. 1999, 7, 217–231. [Google Scholar]
- Sah, J.F.; Eckert, R.L.; Chandraratna, R.A.; Rorke, E.A. Retinoids suppress epidermal growth factor-associated cell proliferation by inhibiting epidermal growth factor receptor-dependent ERK1/2 activation. J. Biol. Chem. 2002, 277, 9728–9735. [Google Scholar] [CrossRef]
- Hochmann, J.; Sobrinho, J.S.; Villa, L.L.; Sichero, L. The Asian-American variant of human papillomavirus type 16 exhibits higher activation of MAPK and PI3K/AKT signaling pathways, transformation, migration and invasion of primary human keratinocytes. Virology 2016, 492, 145–154. [Google Scholar] [CrossRef]
- DuShane, J.K.; Wilczek, M.P.; Crocker, M.A.; Maginnis, M.S. High-Throughput Characterization of Viral and Cellular Protein Expression Patterns During JC Polyomavirus Infection. Front. Microbiol. 2019, 10, 783. [Google Scholar] [CrossRef] [PubMed]
- Shane, J.K.; Maginnis, M.S. Human DNA Virus Exploitation of the MAPK-ERK Cascade. Int. J. Mol. Sci. 2019, 20, 3427. [Google Scholar]
- Kim, M.K.; Kim, H.S.; Kim, S.H.; Oh, J.M.; Han, J.Y.; Lim, J.M.; Juhnn, Y.S.; Song, Y.S. Human papillomavirus type 16 E5 oncoprotein as a new target for cervical cancer treatment. Biochem. Pharmacol. 2010, 80, 1930–1935. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lin, B.; Liu, X.; Zhang, W.; Zhang, E.; Hu, L.; Ma, Y.; Li, X.; Tang, X. ERK Signaling Pathway Is Involved in HPV-16 E6 but not E7 Oncoprotein-Induced HIF-1α Protein Accumulation in NSCLC Cells. Oncol. Res. 2016, 23, 109–118. [Google Scholar] [CrossRef]
- Hays, E.; Bonavida, B. YY1 regulates cancer cell immune resistance by modulating PD-L1 expression. Drug Resist. Updates 2019, 43, 10–28. [Google Scholar] [CrossRef]
- Di Croce, L.; Helin, K. Transcriptional regulation by Polycomb group proteins. Nat. Struct. Mol. Biol. 2013, 20, 1147–1155. [Google Scholar] [CrossRef]
- Gordon, S.; Akopyan, G.; Garban, H.; Bonavida, B. Transcription factor YY1: Structure, function, and therapeutic implications in cancer biology. Oncogene 2006, 25, 1125–1142. [Google Scholar] [CrossRef]
- Sui, G. The Regulation of YY1 in Tumorigenesis and its Targeting Potential in Cancer Therapy. Mol. Cell. Pharmacol. 2009, 1, 157–176. [Google Scholar] [CrossRef]
- Bauknecht, T.; Shi, Y. Overexpression of C/EBPbeta represses human papillomavirus type 18 upstream regulatory region activity in HeLa cells by interfering with the binding of TATA-binding protein. J. Virol. 1998, 72, 2113–2124. [Google Scholar] [CrossRef]
- Bushmeyer, S.; Park, K.; Atchison, M.L. Characterization of functional domains within the multifunctional transcription factor, YY1. J. Biol. Chem. 1995, 270, 30213–30220. [Google Scholar] [CrossRef]
- Lichy, J.H.; Majidi, M.; Elbaum, J.; Tsai, M.M. Differential expression of the human ST5 gene in HeLa-fibroblast hybrid cell lines mediated by YY1: Evidence that YY1 plays a part in tumor suppression. Nucleic Acids Res. 1996, 24, 4700–4708. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, C.C.; Chen, J.J.; Yang, P.C. Multifunctional transcription factor YY1: A therapeutic target in human cancer? Expert Opin. Ther. Targets 2006, 10, 253–266. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Wang, Q.; Zhou, Y.; Wu, X.; Wang, L.; Duru, N.; Kong, X.; Zhang, P.; Wan, B.; Sui, L.; et al. YY1 Is a Novel Potential Therapeutic Target for the Treatment of HPV Infection-Induced Cervical Cancer by Arsenic Trioxide. Int. J. Gynecol. Cancer 2011, 21, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef] [PubMed]
- Cha, D.; O’Brien, P.; O’Toole, E.A.; Woodley, D.T.; Hudson, L.G. Enhanced Modulation of Keratinocyte Motility by Transforming Growth Factor-α (TGF-α) Relative to Epidermal Growth Factor (EGF). J. Investig. Dermatol. 1996, 106, 590–597. [Google Scholar] [CrossRef]
- Yarden, Y. The EGFR family and its ligands in human cancer: Signalling mechanisms and therapeutic opportunities. Eur. J. Cancer 2001, 37, 3–8. [Google Scholar] [CrossRef]
- Franklin, W.A.; Veve, R.; Hirsch, F.R.; Helfrich, B.A.; Bunn, P.A. Epidermal growth factor receptor family in lung cancer and premalignancy. Semin. Oncol. 2002, 29, 3–14. [Google Scholar] [CrossRef]
- Hirsch, F.R.; Scagliotti, G.V.; Langer, C.J.; Varella-Garcia, M.; Franklin, W.A. Epidermal growth factor family of receptors in preneoplasia and lung cancer: Perspectives for targeted therapies. Lung Cancer 2003, 41, 29–42. [Google Scholar] [CrossRef]
- Makinoshima, H.; Takita, M.; Matsumoto, S.; Yagishita, A.; Owada, S.; Esumi, H.; Tsuchihara, K. Epidermal Growth Factor Receptor (EGFR) Signaling Regulates Global Metabolic Pathways in EGFR-mutated Lung Adenocarcinoma. J. Biol. Chem. 2014, 289, 20813–20823. [Google Scholar] [CrossRef]
- Babic, I.; Anderson, E.S.; Tanaka, K.; Guo, D.; Masui, K.; Li, B.; Zhu, S.; Gu, Y.; Villa, G.R.; Akhavan, D.; et al. EGFR Mutation-Induced Alternative Splicing of Max Contributes to Growth of Glycolytic Tumors in Brain Cancer. Cell Metab. 2013, 17, 1000–1008. [Google Scholar] [CrossRef]
- Makinoshima, H.; Takita, M.; Saruwatari, K.; Umemura, S.; Obata, Y.; Matsumoto, S.; Sugiyama, E.; Ochiai, A.; Abe, R.; Goto, K.; et al. Signaling through the phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) axis is responsible for aerobic glycolysis mediated by glucose transporter in epidermal growth factor receptor (EGFR)-mutated lung adenocarcinoma. J. Biol. Chem. 2015, 290, 17495–17504. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, D.; Stellwag, B.; Borlinghaus, P.; Meier, W.; Scheidel, P. Clinical implications of the epidermal growth factor receptor in the squamous cell carcinoma of the uterine cervix. Gynecol. Oncol. 1989, 33, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Ilahi, N.E.; Bhatti, A. Impact of HPV E5 on viral life cycle via EGFR signaling. Microb. Pathog. 2020, 139, 103923. [Google Scholar] [CrossRef]
- Kim, S.-H.; Juhnn, Y.-S.; Kang, S.; Park, S.-W.; Sung, M.-W.; Bang, Y.-J.; Song, Y.-S. Human papillomavirus 16 E5 up-regulates the expression of vascular endothelial growth factor through the activation of epidermal growth factor receptor, MEK/ ERK1,2 and PI3K/Akt. Cell. Mol. Life Sci. 2006, 63, 930–938. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Oh, J.M.; No, J.H.; Bang, Y.J.; Juhnn, Y.S.; Song, Y.S. Involvement of NF-κB and AP-1 in COX-2 upregulation by human papillomavirus 16 E5 oncoprotein. Carcinogenesis 2009, 30, 753–757. [Google Scholar] [CrossRef] [PubMed]
- Straight, S.W.; Hinkle, P.M.; Jewers, R.J.; McCance, D.J. The E5 oncoprotein of human papillomavirus type 16 transforms fibroblasts and effects the downregulation of the epidermal growth factor receptor in keratinocytes. J. Virol. 1993, 67, 4521–4532. [Google Scholar] [CrossRef]
- Crusius, K.; Auvinen, E.; Steuer, B.; Gaissert, H.; Alonso, A. The human papillomavirus type 16 E5-protein modulates ligand-dependent activation of the EGF receptor family in the human epithelial cell line HaCaT. Exp. Cell Res. 1998, 241, 76–83. [Google Scholar] [CrossRef]
- Dassonville, O.; Formento, J.L.; Francoual, M.; Ramaioli, A.; Santini, J.; Schneider, M.; Demard, F.; Milano, G. Expression of epidermal growth factor receptor and survival in upper aerodigestive tract cancer. J. Clin. Oncol. 1993, 11, 1873–1878. [Google Scholar] [CrossRef]
- Sheridan, M.T.; O’Dwyer, T.; Seymour, C.B.; Mothersill, C.E. Potential indicators of radiosensitivity in squamous cell carcinoma of the head and neck. Radiat. Oncol. Investig. 1997, 5, 180–186. [Google Scholar] [CrossRef]
- Balaban, N.; Moni, J.; Shannon, M.; Dang, L.; Murphy, E.; Goldkorn, T. The effect of ionizing radiation on signal transduction: Antibodies to EGF receptor sensitize A431 cells to radiation. Biochim. Biophys. Acta Mol. Cell Res. 1996, 1314, 147–156. [Google Scholar] [CrossRef]
- Valle-Mendiola, A.; Bustos-Rodríguez, R.; Domínguez-Melendez, V.; Zerecero-Carreón, O.; Gutiérrez-Hoya, A.; Weiss-Steider, B.; Soto-Cruz, I. Mutations in the helix αC of the catalytic domain from the EGFR affect its activity in cervical cancer cell lines. Oncol. Lett. 2022, 23, 13191. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, E.; Prowse, D.M.; Ng, M.; Cuzick, J.; Mesher, D.; Hiscock, F.; Lu, Y.-J.; Watkin, N.; Corbishley, C.; Lam, W.; et al. Alternative HER/PTEN/Akt Pathway Activation in HPV Positive and Negative Penile Carcinomas. PLoS ONE 2011, 6, e17517. [Google Scholar] [CrossRef] [PubMed]
- Mazibrada, J.; Longo, L.; Vatrano, S.; Cappia, S.; Giorcelli, J.; Pentenero, M.; Gandolfo, S.; Volante, M.; Dell’Oste, V.; Cigno, I.L.; et al. Differential expression of HER2, STAT3, SOX2, IFI16 and cell cycle markers during HPV-related head and neck carcinogenesis. New Microbiol. 2014, 37, 129–143. [Google Scholar] [PubMed]
- Perez-Regadera, J.; Sanchez-Munoz, A.; De-la-Cruz, J.; Ballestin, C.; Lora, D.; Garcia-Martin, R. Negative prognostic impact of the coexpression of epidermal growth factor receptor and c-erbB-2 in locally advanced cervical cancer. Oncology 2009, 76, 133–141. [Google Scholar] [CrossRef]
- Ojesina, A.I.; Lichtenstein, L.; Freeman, S.S.; Pedamallu, C.S.; Imaz-Rosshandler, I.; Pugh, T.J.; Cherniack, A.D.; Ambrogio, L.; Cibulskis, K.; Bertelsen, B.; et al. Landscape of genomic alterations in cervical carcinomas. Nature 2014, 506, 371–375. [Google Scholar] [CrossRef]
- Pollock, N.I.; Wang, L.; Wallweber, G.; Gooding, W.E.; Huang, W.; Chenna, A.; Winslow, J.; Sen, M.; DeGrave, K.A.; Li, H.; et al. Increased Expression of HER2, HER3, and HER2:HER3 Heterodimers in HPV-Positive HNSCC Using a Novel Proximity-Based Assay: Implications for Targeted Therapies. Clin. Cancer Res. 2015, 21, 4597–4606. [Google Scholar] [CrossRef]
- Tilborghs, S.; Corthouts, J.; Verhoeven, Y.; Arias, D.; Rolfo, C.; Trinh, X.B.; van Dam, P.A. The role of Nuclear Factor-kappa B signaling in human cervical cancer. Crit. Rev. Oncol. Hematol. 2017, 120, 141–150. [Google Scholar] [CrossRef]
- Hayden, M.S.; West, A.P.; Ghosh, S. NF-kappaB and the immune response. Oncogene 2006, 25, 6758–6780. [Google Scholar] [CrossRef]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef]
- Lin, Y.; Bai, L.; Chen, W.; Xu, S. The NF-κB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert Opin. Ther. Targets 2009, 14, 45–55. [Google Scholar] [CrossRef]
- Branca, M.; Giorgi, C.; Ciotti, M.; Santini, D.; Di Bonito, L.; Costa, S.; Benedetto, A.; Bonifacio, D.; Di Bonito, P.; Paba, P.; et al. Upregulation of nuclear factor-kappaB (NF-kappaB) is related to the grade of cervical intraepithelial neoplasia, but is not an independent predictor of high-risk human papillomavirus or disease outcome in cervical cancer. Diagn. Cytopathol. 2006, 34, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Bharti, A.C.; Varghese, P.; Saluja, D.; Das, B.C. Differential expression and activation of NF-kappaB family proteins during oral carcinogenesis: Role of high risk human papillomavirus infection. Int. J. Cancer 2006, 119, 2840–2850. [Google Scholar] [CrossRef]
- Gupta, S.; Kumar, P.; Das, B.C. HPV+ve/−ve oral-tongue cancer stem cells: A potential target for relapse-free therapy. Transl. Oncol. 2020, 14, 100919. [Google Scholar] [CrossRef] [PubMed]
- Byg, L.M.; Vidlund, J.; Vasiljevic, N.; Clausen, D.; Forslund, O.; Norrild, B. NF-κB signalling is attenuated by the E7 protein from cutaneous human papillomaviruses. Virus Res. 2012, 169, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Katzenellenbogen, R.A.; Grandori, C.; Galloway, D.A. NFX1 Plays a Role in Human Papillomavirus Type 16 E6 Activation of NFκB Activity. J. Virol. 2010, 84, 11461–11469. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Mo, D.; Liu, H.; Veena, M.S.; Srivatsan, E.S.; Massoumi, R.; Rettig, M.B. Inactivation of the CYLD Deubiquitinase by HPV E6 Mediates Hypoxia-Induced NF-κB Activation. Cancer Cell 2008, 14, 394–407. [Google Scholar] [CrossRef] [PubMed]
- Guttridge, D.C.; Albanese, C.; Reuther, J.Y.; Pestell, R.G.; Baldwin, A.S. NF-κB Controls Cell Growth and Differentiation through Transcriptional Regulation of Cyclin D1. Mol. Cell. Biol. 1999, 19, 5785–5799. [Google Scholar] [CrossRef]
- Rebhandl, S.; Huemer, M.; Greil, R.; Geisberger, R. AID/APOBEC deaminases and cancer. Oncoscience 2015, 2, 320–333. [Google Scholar] [CrossRef]
- Lajer, C.B.; Garnæs, E.; Friis-Hansen, L.; Norrild, B.; Therkildsen, M.H.; Glud, M.; Rossing, M.; Lajer, H.; Svane, D.; Skotte, L.; et al. The role of miRNAs in human papilloma virus (HPV)-associated cancers: Bridging between HPV-related head and neck cancer and cervical cancer. Br. J. Cancer 2012, 106, 1526–1534. [Google Scholar] [CrossRef]
- Shishodia, G.; Shukla, S.; Srivastava, Y.; Masaldan, S.; Mehta, S.; Bhambhani, S.; Sharma, S.; Mehrotra, R.; Das, B.C.; Bharti, A.C. Alterations in microRNAs miR-21 and let-7a correlate with aberrant STAT3 signaling and downstream effects during cervical carcinogenesis. Mol. Cancer 2015, 14, 116. [Google Scholar] [CrossRef]
- Wang, X.; Tang, S.; Le, S.-Y.; Lu, R.; Rader, J.S.; Meyers, C.; Zheng, Z.-M. Aberrant Expression of Oncogenic and Tumor-Suppressive MicroRNAs in Cervical Cancer Is Required for Cancer Cell Growth. PLoS ONE 2008, 3, e2557. [Google Scholar] [CrossRef]
- Božinović, K.; Sabol, I.; Dediol, E.; Gašperov, N.M.; Manojlović, S.; Vojtechova, Z.; Tachezy, R.; Grce, M. Genome-wide miRNA profiling reinforces the importance of miR-9 in human papillomavirus associated oral and oropharyngeal head and neck cancer. Sci. Rep. 2019, 9, 2306. [Google Scholar] [CrossRef] [PubMed]
- Zeng, K.; Zhang, W.; Hu, X. Progress of research in miR-218 and cervical cancer. Chin. Ger. J. Clin. Oncol. 2013, 12, 399–402. [Google Scholar] [CrossRef]
- Martinez, I.; Gardiner, A.S.; Board, K.F.; Monzon, F.A.; Edwards, R.P.; Khan, S.A. Human papillomavirus type 16 reduces the expression of microRNA-218 in cervical carcinoma cells. Oncogene 2008, 27, 2575–2582. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Hu, Y.; Ye, F.; Li, Y.; Lv, W.; Xie, X. Reduced miR-34a Expression in Normal Cervical Tissues and Cervical Lesions with High-Risk Human Papillomavirus Infection. Int. J. Gynecol. Cancer 2010, 20, 597–604. [Google Scholar] [CrossRef]
- Zhang, R.; Su, J.; Xue, S.L.; Yang, H.; Ju, L.L.; Ji, Y.; Wu, K.H.; Zhang, Y.W.; Zhang, Y.X.; Hu, J.F.; et al. HPV E6/p53 mediated down-regulation of miR-34a inhibits Warburg effect through targeting LDHA in cervical cancer. Am. J. Cancer Res. 2016, 6, 312–320. [Google Scholar]
- Sannigrahi, M.K.; Sharma, R.; Singh, V.; Panda, N.K.; Rattan, V.; Khullar, M. Role of host miRNA Hsa-miR-139-3p in HPV-16–induced carcinomas. Clin. Cancer Res. 2017, 23, 3884–3895. [Google Scholar] [CrossRef]




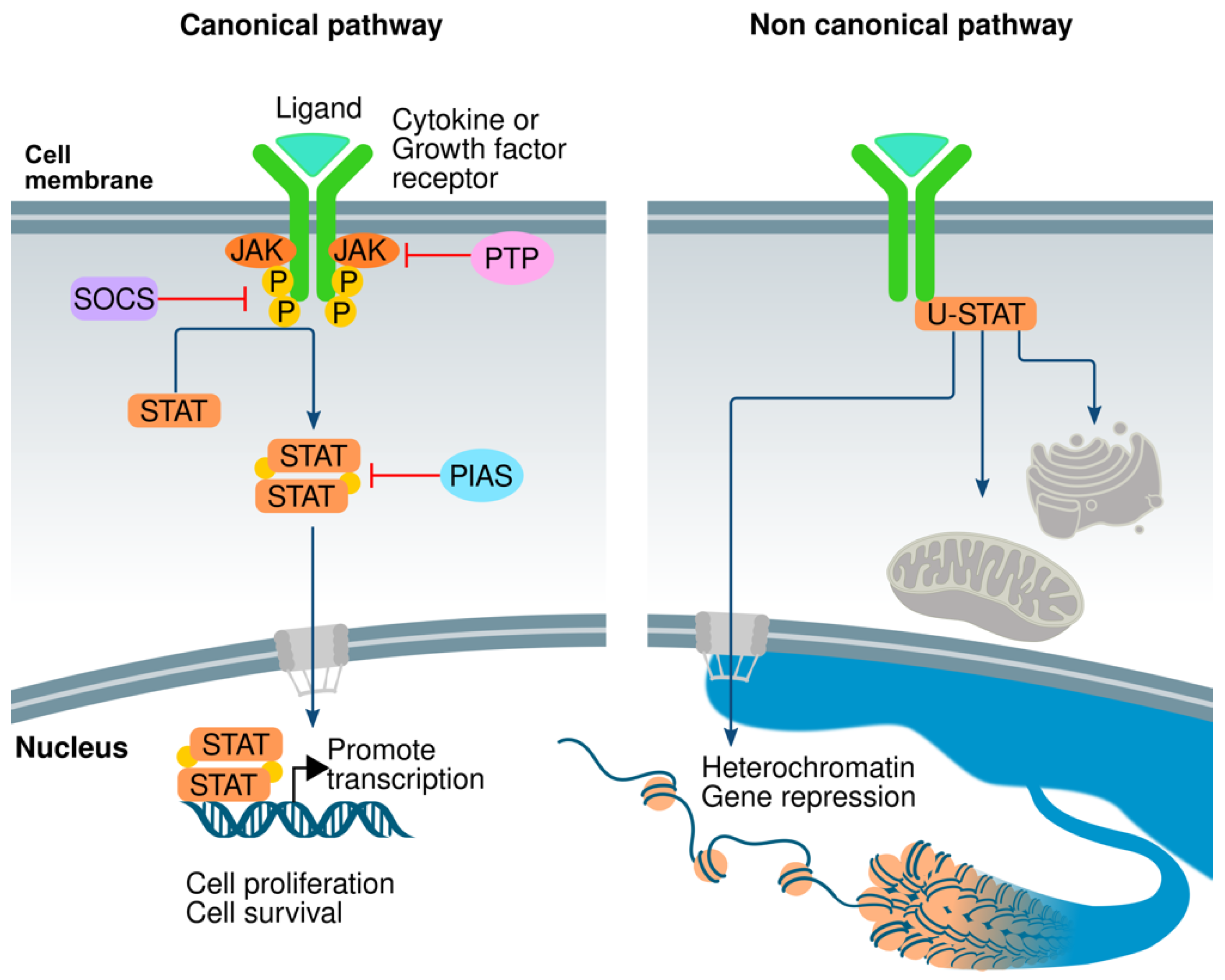{kind=link}
{kind=link}
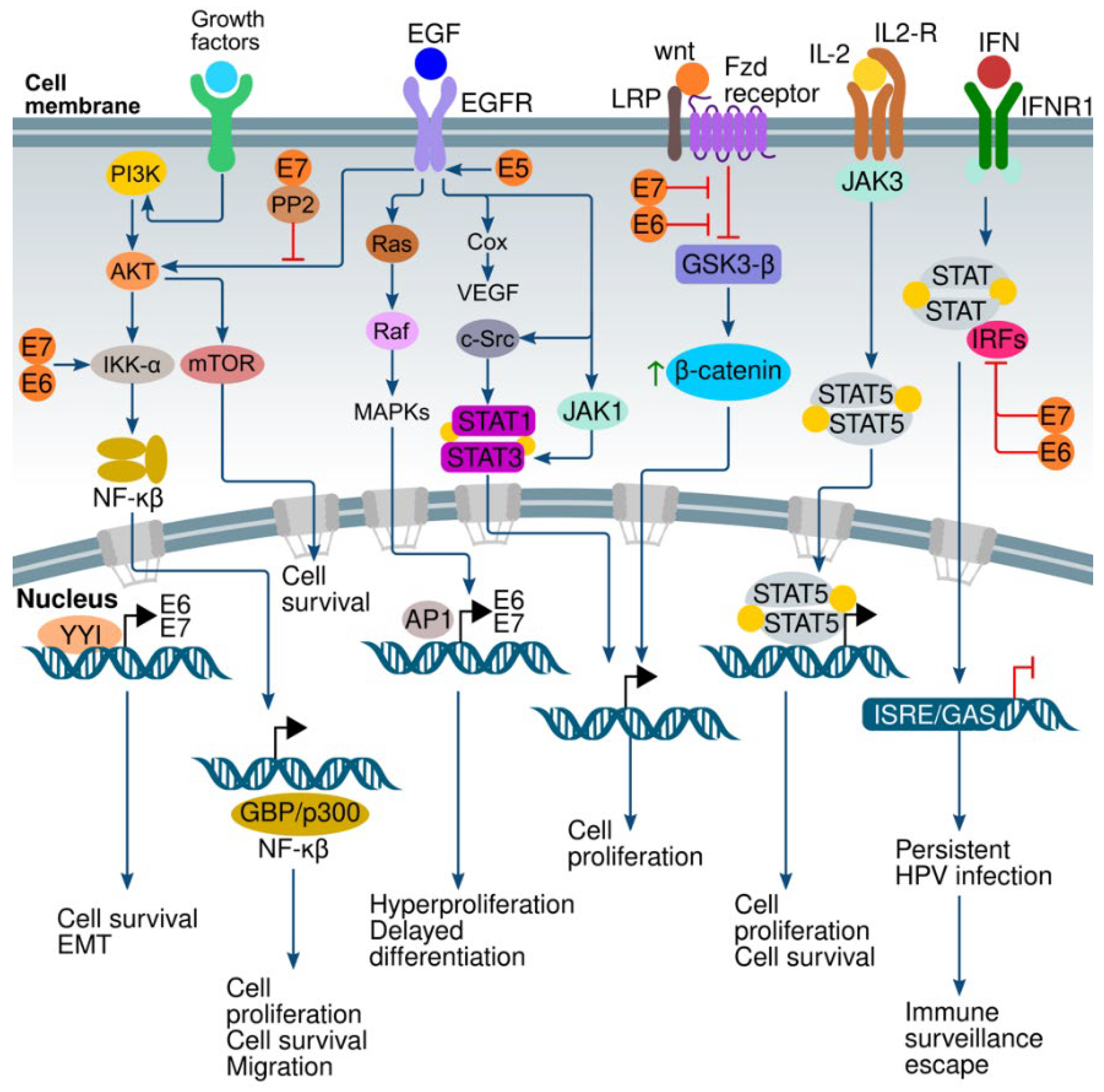{kind=link}
| DBD Sequence (Protein) | Optimal Binding Sequence (DNA) [131] | |
|---|---|---|
| STAT1 | 317-FVVERQPCMPTHPQRPLVLKTGVQFTVKLRLLVKLQELNYNLKVKVLFDKDVNERNTV KGFRKFNILGTHTKVMNMEESTNGSLAAEFRHLQLKEQKNAGTRTNEGPLIVTEELHSL SFETQLCQPGLVIDLETTSLPVVVISNVSQLPSGWASILWYNML-437 | TTCCCGTA |
| STAT2 | 312-FVVETQPCMPQTPHRPLILKTGSKFTVRTRLLVRLQEGNESLTVEVSIDRNPPQL QGFRKFNILTSNQKTLTPEKGQSQGLIWDFGYLTLVEQRSGGSGKGSNKGPLGV TEELHIISFTVKYTYQGLKQELKTDTLPVVIISNMNQLSIAWASVLWFNLL-432 | GGGAAACCGAAACTG |
| STAT3 | 321-FVVERQPCMPMHPDRPLVIKTGVQFTTKVRLLVKFPELNYQLKIKVCIDKDSGD VAALRGSRKFNILGTNTKVMNMEESNNGSLSAEFKHLTLREQRCGNGGRANC DASLIVTEELHLITFETEVYHQGLKIDLETHSLPVVVISNICQMPNAWASILWYN MLT-421 | TTCCCGTAA |
| STAT4 | 316-FVVERQPCMPTHPQRPLVLKTLIQFTVKLRLLIKLPELNYQVKVKASIDKNVSTL SNRRFVLCGTNVKAMSIEESSNGSLSVEFRHLQPKEMKSSAGGKGNEGCHMVT EELHSITFETQICLYGLTIDLETSSLPVVMISNVSQLPNAWASIIWYNVS-436 | TTCCCAGAA |
| STAT5 | 332-FIIEKQPPQVLKTQTKFAATVRLLVGGKLNVHMNPPQVKATIISEQQAKSLLKN ENTRNECSGEILNNCCVMEYHQATGTLSAHFRNMSLKRIKRADRRGAESVTEE KFTVLFESQFSVGSNELVFQVKTLSLPVVVIVHGSQDHNATATVLWDNAFA-452 | TTCCTGGAA |
| STAT6 | 273-FLVEKQPPQVLKTQTKFQAGVRFLLGLRFLGAPAKPPLVRADMVTEKQARELS VPQGPGAESTGEIINNTVPLENSIPGNCCSALFKNLLLKKIKRCERKGTESVTEEK CAVLFSASFTLGPGKLPIQLQALSLPLVVIVHGNQDNNAKATILWDNAF-393 | TTCTGGAA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valle-Mendiola, A.; Gutiérrez-Hoya, A.; Soto-Cruz, I. JAK/STAT Signaling and Cervical Cancer: From the Cell Surface to the Nucleus. Genes 2023, 14, 1141. https://doi.org/10.3390/genes14061141
Valle-Mendiola A, Gutiérrez-Hoya A, Soto-Cruz I. JAK/STAT Signaling and Cervical Cancer: From the Cell Surface to the Nucleus. Genes. 2023; 14(6):1141. https://doi.org/10.3390/genes14061141
Chicago/Turabian StyleValle-Mendiola, Arturo, Adriana Gutiérrez-Hoya, and Isabel Soto-Cruz. 2023. "JAK/STAT Signaling and Cervical Cancer: From the Cell Surface to the Nucleus" Genes 14, no. 6: 1141. https://doi.org/10.3390/genes14061141
APA StyleValle-Mendiola, A., Gutiérrez-Hoya, A., & Soto-Cruz, I. (2023). JAK/STAT Signaling and Cervical Cancer: From the Cell Surface to the Nucleus. Genes, 14(6), 1141. https://doi.org/10.3390/genes14061141