
Development and Application of Real-Time PCR-Based Screening for Identification of Omicron SARS-CoV-2 Variant Sublineages

 ,
,  ,
,  , , , , , , , , , and
, , , , , , , , , and
Abstract
:1. Introduction
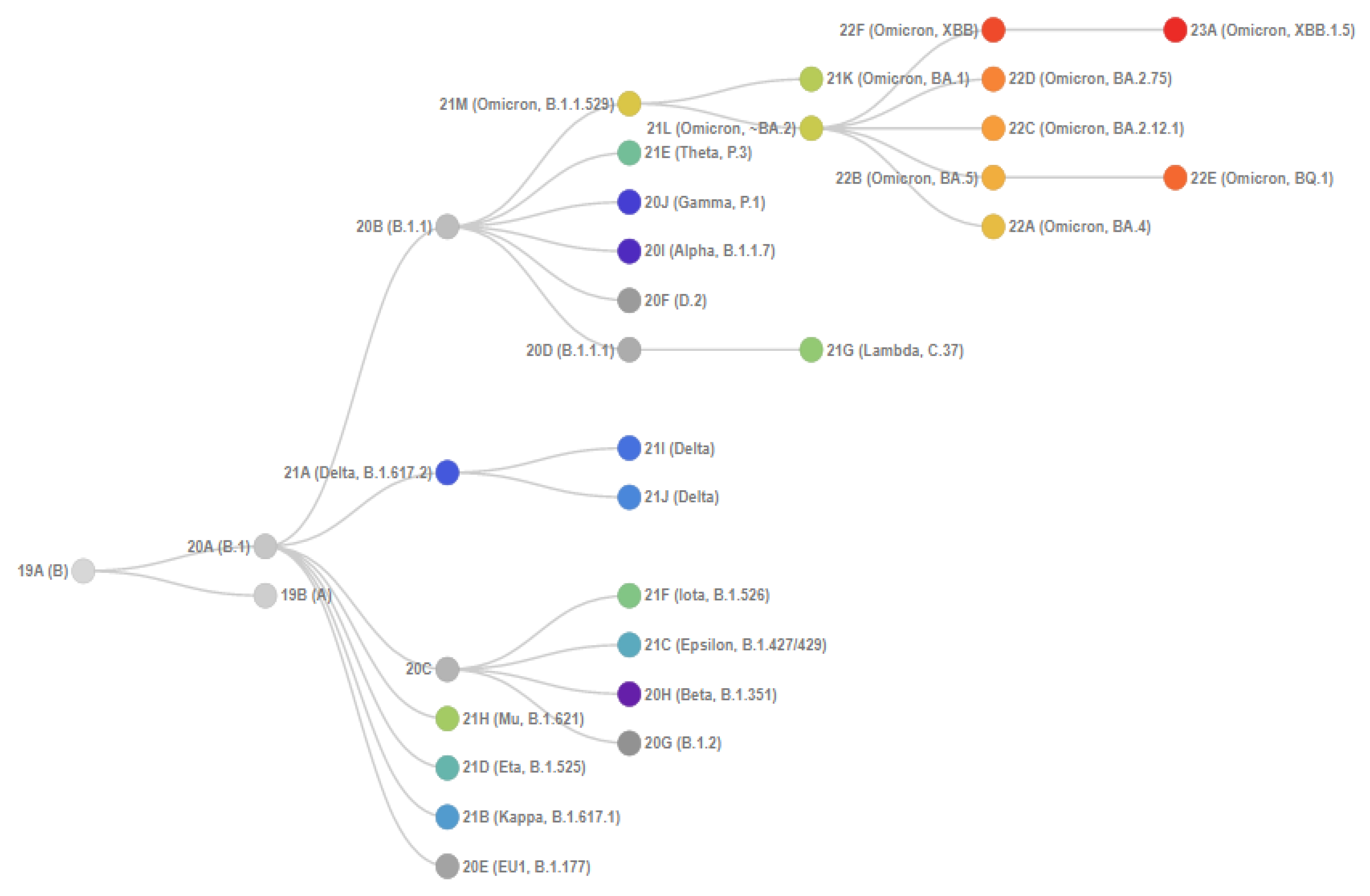
2. Materials and Methods
2.1. Biological Material Samples
2.2. Whole-Genome and Sanger Sequencing Conditions
2.3. Data Analysis
2.4. Real-Time PCR Target Selection
2.5. Development of Multiplex Reverse Transcription-Polymerase Chain Reaction (RT-PCR) for Genotyping of Omicron SARS-CoV-2 Sublineages
2.6. Primer Synthesis for PCR Techniques
2.7. Statistical Analysis
3. Results
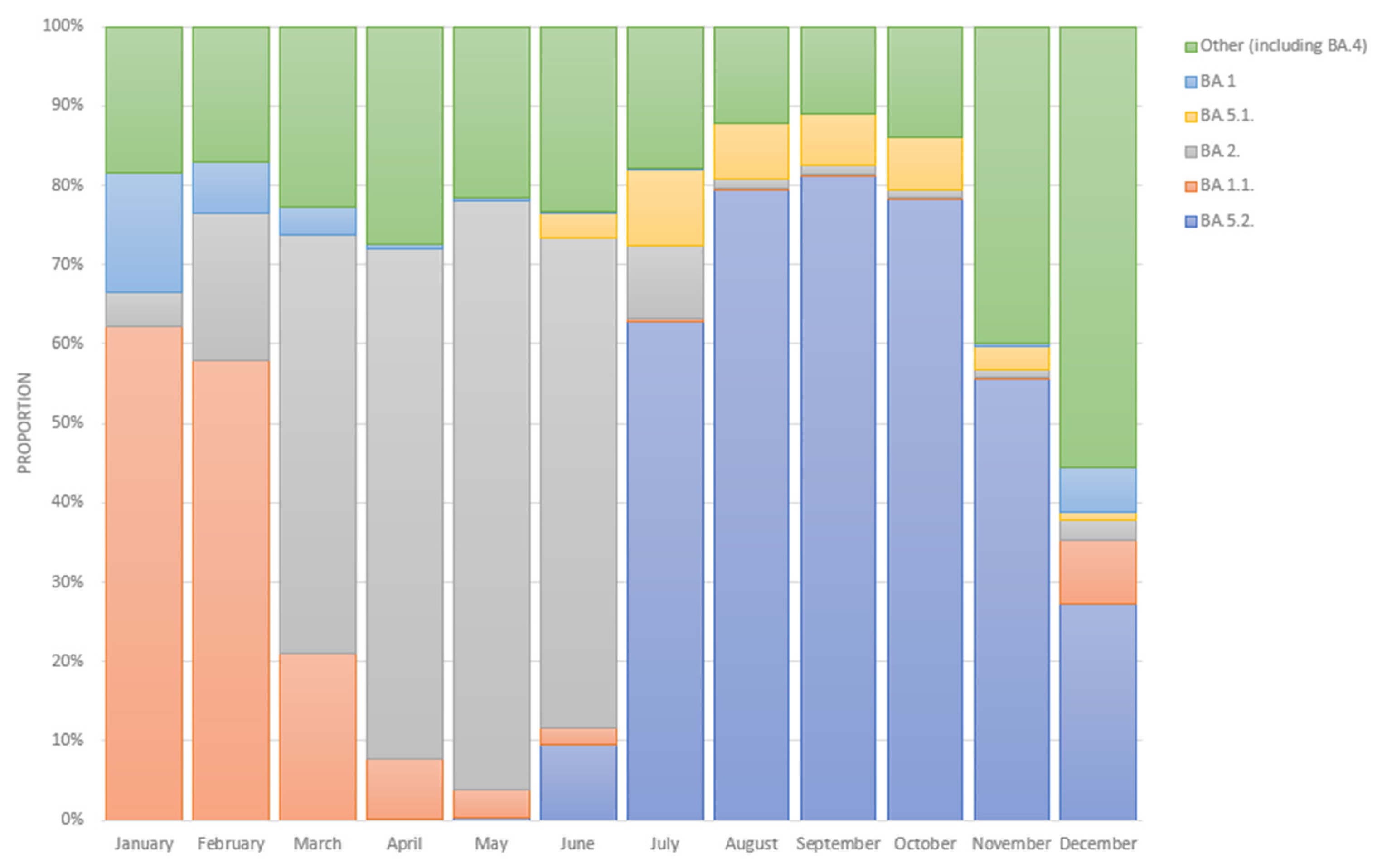
3.1. Epidemiological Analysis of the Prevalence of Omicron Sublineages in Russia
3.2. Results Obtained by Using One-Step Multiplex PCR
4. Discussion
5. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Spratt, A.N.; Kannan, S.R.; Sharma, K.; Sachdev, S.; Kandasamy, S.L.; Sönnerborg, A.; Lorson, C.L.; Singh, K. Continued Complexity of Mutations in Omicron Sublineages. Biomedicines 2022, 10, 2593. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Wang, L.; Zhu, Y.; Lu, L.; Jiang, S. Origin, virological features, immune evasion and intervention of SARS-CoV-2 Omicron sublineages. Signal Transduct. Target. Ther. 2022, 7, 241. [Google Scholar] [CrossRef] [PubMed]
- Tegally, H.; Moir, M.; Everatt, J.; Giovanetti, M.; Scheepers, C.; Wilkinson, E.; Subramoney, K.; Moyo, S.; Amoako, D.G.; Baxter, C.; et al. Continued Emergence and Evolution of Omicron in South Africa: New BA.4 and BA.5 Lineages. MedRxiv 2022. [Google Scholar] [CrossRef]
- Shrestha, L.B.; Foster, C.; Rawlinson, W.; Tedla, N.; Bull, R.A. Evolution of the SARS-CoV-2 omicron variants BA.1 to BA.5: Implications for immune escape and transmission. Rev. Med Virol. 2022, 32, e2381. [Google Scholar] [CrossRef] [PubMed]
- CoVariants. Available online: https://Covariants.Org/ (accessed on 8 March 2023).
- Real-Time Tracking of Pathogen Evolution, (Nextrain). Available online: https://Nextstrain.Org/ (accessed on 8 March 2023).
- Gwinn, M.; MacCannell, D.; Armstrong, G.L. Next-Generation Sequencing of Infectious Pathogens. JAMA 2019, 321, 893–894. [Google Scholar] [CrossRef] [Green Version]
- Akimkin, V.G.; Popova, A.Y.; Khafizov, K.F.; Dubodelov, D.V.; Ugleva, S.V.; Semenenko, T.A.; Ploskireva, A.A.; Gorelov, A.V.; Pshenichnaya, N.Y.; Yezhlova, E.; et al. COVID-19: Evolution of the pandemic in Russia. Report II: Dynamics of the circulation of SARS-CoV-2 genetic variants. J. Microbiol. Epidemiol. Immunobiol. 2022, 99, 381–396. [Google Scholar] [CrossRef]
- Global Initiative on Sharing Avian Influenza Data (GISAID). Available online: Https://Gisaid.Org/ (accessed on 8 March 2023).
- Lai, E.; Kennedy, E.B.; Lozach, J.; Hayashibara, K.; Davis-Turak, J.; Becker, D.; Brzoska, P.; Cassens, T.; Diamond, E.; Gandhi, M.; et al. A Method for Variant Agnostic Detection of SARS-CoV-2, Rapid Monitoring of Circulating Variants, and Early Detection of Emergent Variants Such as Omicron. J. Clin. Microbiol. 2022, 60, e00342-22. [Google Scholar] [CrossRef]
- Akimkin, V.G.; Semenenko, T.A.; Ugleva, S.V.; Dubodelov, D.V.; Kuzin, S.N.; Yacyshina, S.B.; Khafizov, K.F.; Petrov, V.V.; Cherkashina, A.S.; Gasanov, G.A.; et al. COVID-19 in Russia: Epidemiology and Molecular Genetic Monitoring. Ann. Russ. Acad. Med Sci. 2022, 77, 254–260. [Google Scholar] [CrossRef]
- Neopane, P.; Nypaver, J.; Shrestha, R.; Beqaj, S.S. SARS-CoV-2 Variants Detection Using TaqMan SARS-CoV-2 Mutation Panel Molecular Genotyping Assays. Infect. Drug Resist. 2021, 14, 4471–4479. [Google Scholar] [CrossRef]
- Handbook of Biomaterials for PCR-Diagnostics of the Central Research Institute of Epidemiology of FSSCRP Human Wellbeing. Available online: https://Prepcr.Crie.Ru/ (accessed on 8 March 2023).
- Kotov, I.; Saenko, V.; Borisova, N.; Kolesnikov, A.; Kondrasheva, L.; Tivanova, E.; Khafizov, K.; Akimkin, V. Effective Approaches to Study the Genetic Variability of SARS-CoV-2. Viruses 2022, 14, 1855. [Google Scholar] [CrossRef]
- Cherkashina, A.S. SARS-CoV-2 S-Gene Sanger Sequencing. protocols.io. Available online: https://dx.doi.org/10.17504/protocols.io.3byl4jnwzlo5/v1 (accessed on 30 May 2023).
- Borisova, N.I.; Kotov, I.A.; Kolesnikov, A.A.; Kaptelova, V.V.; Speranskaya, A.S.; Kondrasheva, L.Y.; Tivanova, E.V.; Khafizov, K.F.; Akimkin, V.G. Monitoring the spread of the SARS-CoV-2 (Coronaviridae: Coronavirinae: Betacoronavirus; Sarbecovirus) variants in the Moscow region using targeted high-throughput sequencing. Probl. Virol. 2021, 66, 269–278. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Virus Genome Aggregator of Russia (VGARus). Available online: Https://Genome.Crie.Ru/ (accessed on 8 March 2023).
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of Epidemiological Lineages in an Emerging Pandemic Using the Pangolin Tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef] [PubMed]
- Pango Lineages: Latest Epidemiological Lineages of SARS-CoV-2. Available online: https://Cov-Lineages.Org/ (accessed on 8 March 2023).
- Flowchart Maker & Online Diagram Software. Available online: https://App.Diagrams.Net/ (accessed on 24 November 2022).
- Hall, T.; Biosciences, I.; Carlsbad, C.J.G.B.B. BioEdit: An Important Software for Molecular Biology. GERF Bull. Biosci. 2011, 2, 60–61. [Google Scholar]
- Hall, T.A. BioEdit: A User Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Khare, S.; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; Ho, J.; Lee, R.T.; Yeo, W.; et al. GISAID’s Role in Pandemic Response. China CDC Wkly. 2021, 3, 1049–1051. [Google Scholar] [CrossRef]
- Gangavarapu, K.; Latif, A.A.; Mullen, J.L.; Alkuzweny, M.; Hufbauer, E.; Tsueng, G.; Haag, E.; Zeller, M.; Aceves, C.M.; Zaiets, K.; et al. Outbreak. Info Genomic Reports: Scalable and Dynamic Surveillance of SARS-CoV-2 Variants and Mutations. Nat. Methods 2022, 20, 512–522. [Google Scholar] [CrossRef]
- Integrated DNA Technologies. Integrated DNA Technologies∣IDT. Available online: https://Eu.Idtdna.Com/Pages (accessed on 6 February 2023).
- Ellington, A.; Pollard, J.D., Jr. Introduction to the Synthesis and Purification of Oligonucleotides. Curr. Protoc. Nucleic Acid Chem. 2000, 1, A.3C.1–A.3C.22. [Google Scholar] [CrossRef]
- Epitools—Calculate Confidence Limits for a Sample Proportion. Available online: Https://Epitools.Ausvet.Com.Au/Ciproportion (accessed on 8 March 2023).
- Brown, L.D.; Cai, T.T.; DasGupta, A. Interval Estimation for a Binomial Proportion. Stat. Sci. 2001, 16, 101–133. [Google Scholar] [CrossRef]
- Esman, A.; Cherkashina, A.; Mironov, K.; Dubodelov, D.; Salamaikina, S.; Golubeva, A.; Gasanov, G.; Khafizov, K.; Petrova, N.; Cherkashin, E.; et al. SARS-CoV-2 Variants Monitoring Using Real-Time PCR. Diagnostics 2022, 12, 2388. [Google Scholar] [CrossRef]
- The Official Online Resource for Informing the Public about Coronavirus. Available online: https://xn--80aesfpebagmfblc0a.xn--p1ai/ (accessed on 8 March 2023). (In Russian).
- Akimkin, V.G.; Popova, A.Y.; Ploskireva, A.A.; Ugleva, S.V.; Semenenko, T.A.; Pshenichnaya, N.Y.; Ezhlova, E.B.; Letyushev, A.N.; Demina, Y.V.; Kuzin, S.N.; et al. COVID-19: The evolution of the pandemic in Russia. Report I: Manifestations of the COVID-19 epidemic process. J. Microbiol. Epidemiol. Immunobiol. 2022, 99, 269–286. [Google Scholar] [CrossRef]
- Tracking SARS-CoV-2 Variants. Available online: https://www.Who.Int/Activities/Tracking-SARS-CoV-2-Variants (accessed on 20 February 2023).
- Durmaz, B.; Abdulmajed, O.; Durmaz, R. Mutations Observed in the SARS-CoV-2 Spike Glycoprotein and Their Effects in the Interaction of Virus with ACE-2 Receptor. Medeni. Med. J. 2020, 35, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, M.; Ferguson, I.D.; Miao, W.; Khavari, P.A. SARS-CoV-2 B.1.1.7 and B.1.351 spike variants bind human ACE2 with increased affinity. Lancet Infect. Dis. 2021, 21, 1070. [Google Scholar] [CrossRef] [PubMed]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Consortium, C.-G.U.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Pondé, R. Physicochemical effect of the N501Y, E484K/Q, K417N/T, L452R and T478K mutations on the SARS-CoV-2 spike protein RBD and its influence on agent fitness and on attributes developed by emerging variants of concern. Virology 2022, 572, 44–54. [Google Scholar] [CrossRef]
- Novazzi, F.; Baj, A.; Genoni, A.; Focosi, D.; Maggi, F. Expansion of L452R-Positive SARS-CoV-2 Omicron Variant, Northern Lombardy, Italy. Emerg. Infect. Dis. 2022, 28, 1301. [Google Scholar] [CrossRef]
- Auspice. Available online: https://Nextstrain.Org/Ncov/Gisaid/Global/6m (accessed on 8 March 2023).
- CovSPECTRUM. Available online: https://Cov-Spectrum.Org (accessed on 8 March 2023).
- Callaway, E. COVID ‘variant soup’ is making winter surges hard to predict. Nature 2022, 611, 213–214. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Johnson, B.A.; Xia, H.; Ku, Z.; Schindewolf, C.; Widen, S.G.; An, Z.; Weaver, S.C.; Menachery, V.D.; et al. Delta spike P681R mutation enhances SARS-CoV-2 fitness over Alpha variant. Cell Rep. 2022, 39, 110829. [Google Scholar] [CrossRef]
- Greaney, A.J.; Starr, T.N.; Barnes, C.O.; Weisblum, Y.; Schmidt, F.; Caskey, M.; Gaebler, C.; Cho, A.; Agudelo, M.; Finkin, S.; et al. Mapping mutations to the SARS-CoV-2 RBD that escape binding by different classes of antibodies. Nat. Commun. 2021, 12, 4196. [Google Scholar] [CrossRef]
- Greaney, A.J.; Loes, A.N.; Crawford, K.H.; Starr, T.N.; Malone, K.D.; Chu, H.Y.; Bloom, J.D. Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe 2021, 29, 463–476.e6. [Google Scholar] [CrossRef]
- Greaney, A.J.; Starr, T.N.; Gilchuk, P.; Zost, S.J.; Binshtein, E.; Loes, A.N.; Hilton, S.K.; Huddleston, J.; Eguia, R.; Crawford, K.H.; et al. Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition. Cell Host Microbe 2020, 29, 44–57.e9. [Google Scholar] [CrossRef] [PubMed]
- Tiecco, G.; Storti, S.; Arsuffi, S.; Degli Antoni, M.; Focà, E.; Castelli, F.; Quiros-Roldan, E. Omicron BA.2 Lineage, the “Stealth” Variant: Is It Truly a Silent Epidemic? A Literature Review. Int. J. Mol. Sci. 2022, 23, 7315. [Google Scholar] [CrossRef] [PubMed]
- Venkatakrishnan, A.J.; Anand, P.; Lenehan, P.J.; Suratekar, R.; Raghunathan, B.; Niesen, M.J.M.; Soundararajan, V. On the Origins of Omicron’s Unique Spike Gene Insertion. Vaccines 2022, 10, 1509. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cheng, G. Sequence analysis of the emerging SARS-CoV-2 variant Omicron in South Africa. J. Med Virol. 2021, 94, 1728–1733. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SARS-CoV-2 S-Gene Mutations | 5′-Sequence-3′ |
|---|---|
| L452R | (F) GGC TGC GTT ATA GCT TGG AAT TCT |
| (R) CCG GCC TGA TAG ATT TCA GTT GAA (P) * (R6G)AAT TAC CGG TAT AGA T (BHQ1) | |
| N501Y | (F) CTG AAA TCT ATC AGG CCG GTA |
| (R) GCT GGT GCA TGT AGA AGT TCA AAA G (P) * (FAM)CCC ACT TAT GGT G (BHQ1) | |
| T95I | (F) GGT TTG ATA ACC CTG TCC TAC CA |
| (R) GGG ACT GGG TCT TCG AAT CTA A (P) * (ROX)GCT TCC ATT GAG AA (BHQ-2) | |
| delHV69-70 | (F) GGA CTT GTT CTT ACC TTT CTT TTC CAA TG |
| (R) TGG AAG CAA AAT AAA CAC CAT CAT TAA AT (P) * (R6G)TCC ATG CTA TCT CTG GGA C (BHQ1) | |
| delLPP24-26 | (F) CAC TAG TCT CTA GTC AGT GTG T |
| (R) TGT CAG GGT AAT AAA CAC CAC G (P) * (FAM)GAA CTC AAT CAT ACA CT (BHQ-1) | |
| Ins214EPE | (F) GGA CCT TGA AGG AAA ACA GGG TAA (R) CCA ATG GTT CTA AAG CCG AAA AAC C (P) * (ROX)TAG TGC GTG AGC CAG AA (BHQ2) |
| P681R | (F) GTA GGC AAT GAT GGA TTG ACT AGC TAC |
| (R) TGC AGG TAT ATG CGC TAG TTA TCA GA (P) * (R6G)GCC GAC GAG AA (BHQ1) | |
| E484A | (F) TTC AAC TGA AAT CTA TCA GGC CG |
| (R) AGT TGC TGG TGC ATG TAG AAG TTC A (P) * (FAM)GTG TTG CAG GTG (BHQ-1) |
| BA.1 | BA.2 | BA.3 | BA.4/BA.5 | Delta | |
|---|---|---|---|---|---|
| L452R | - | - | - | + | + |
| P681R | - | - | - | - | + |
| N501Y | + | + | + | + | - |
| delHV69-70 | + | - | + | + | - |
| Ins214EPE | + | - | - | - | - |
| E484A | + | + | + | + | - |
| delLPP24-26 | - | + | - | + | - |
| T95I | + | - | + | - | - * |
| N501Y | L452R | T95I | Del LPP24-26 | 69-70 | Ins214 | E484A | P681R | |
|---|---|---|---|---|---|---|---|---|
| NGS/Sanger negative | 10 | 26 | 245 | 22 | 71 | 120 | 16 | 110 |
| True Negative (TN) | 8 | 26 | 241 | 20 | 63 | 120 | 14 | 110 |
| False Positive (FP) | 2 | 0 | 4 | 2 | 8 | 0 | 2 | 0 |
| Specificity/(CI) | 80.0% (49.0–94.4%) * | 100% (87.1–100%) * | 98.4% (95.9–99.6%) | 90.9% (72.2–97.5%) * | 88.7% (79.0–95.0%) | 100% (97.0–100%) | 87.5% (64.0–96.5%) * | 100% (97.4–100%) |
| NGS/Sanger positive | 213 | 192 | 46 | 235 | 276 | 3 | 255 | 0 ** |
| True Positive (TP) | 206 | 183 | 43 | 235 | 270 | 3 | 251 | - |
| False Negative (FN) | 7 | 9 | 3 | 0 | 6 | 0 | 4 | - |
| Sensitivity/(CI) | 96.7% (93.4–98.7%) | 95.3% (91.3–97.8%) | 93.5% (82.1–98.6%) | 100% (98.4–100%) | 97.8% (95.3–99.2%) | 100% (43.9–100%) * | 98.4% (96.0–99.6%) | - |
| Total | 223 | 218 | 291 | 257 | 347 | 123 | 271 | 110 |
| Accuracy | 96.0% (92.5–98.1%) | 95.9% (92.3–98.1%) | 97.6% (95.16–99.06%) | 99.26% (97.26–99.96%) | 96% (93.36–97.8%) | 100% (97.1–100%) | 97.8% (95.2–99.2%) | 100% (96.7–100%) |
| BA.1 | BA.2 | BA.3 | BA.4/5 | Delta | Not Detected | Total | Confidence Level/Margin of Error * | |
|---|---|---|---|---|---|---|---|---|
| July | 2 (4.3%) | 17 (37.0%) | 0 (0.0%) | 27 (58.7%) | 0 (0.0%) | 0 (0.0%) | 46 (100.0%) | 90%/10% |
| August | 0 (0.0%) | 2 (3.8%) | 0 (0.0%) | 51 (96.2%) | 0 (0.0%) | 0 (0.0%) | 53 (100.0%) | 90%/10% |
| September | 0 (0.0%) | 2 (1.4%) | 0 (0.0%) | 139 (98.6%) | 0 (0.0%) | 0 (0.0%) | 141 (100.0%) | 95%/2% |
| October | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 181 (95.3%) | 2 (1.1%) | 7 (3.7%) | 190 (100.0%) | 99%/1% |
| November | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 52 (100.0%) | 0 (0.0%) | 0 (0.0%) | 52 (100.0%) | 90%/10% |
| December | 0 (0.0%) | 3 (6.0%) | 0 (0.0%) | 47 (94.0%) | 0 (0.0%) | 0 (0.0%) | 50 (100.0%) | 90%/10% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esman, A.; Dubodelov, D.; Khafizov, K.; Kotov, I.; Roev, G.; Golubeva, A.; Gasanov, G.; Korabelnikova, M.; Turashev, A.; Cherkashin, E.; et al. Development and Application of Real-Time PCR-Based Screening for Identification of Omicron SARS-CoV-2 Variant Sublineages. Genes 2023, 14, 1218. https://doi.org/10.3390/genes14061218
Esman A, Dubodelov D, Khafizov K, Kotov I, Roev G, Golubeva A, Gasanov G, Korabelnikova M, Turashev A, Cherkashin E, et al. Development and Application of Real-Time PCR-Based Screening for Identification of Omicron SARS-CoV-2 Variant Sublineages. Genes. 2023; 14(6):1218. https://doi.org/10.3390/genes14061218
Chicago/Turabian StyleEsman, Anna, Dmitry Dubodelov, Kamil Khafizov, Ivan Kotov, German Roev, Anna Golubeva, Gasan Gasanov, Marina Korabelnikova, Askar Turashev, Evgeniy Cherkashin, and et al. 2023. "Development and Application of Real-Time PCR-Based Screening for Identification of Omicron SARS-CoV-2 Variant Sublineages" Genes 14, no. 6: 1218. https://doi.org/10.3390/genes14061218