
Case Report—An Inherited Loss-of-Function NRXN3 Variant Potentially Causes a Neurodevelopmental Disorder with Autism Consistent with Previously Described 14q24.3-31.1 Deletions

 , and
, and {kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Whole-Exome Sequencing
2.2. Web Resources/Tools and Databases Used for the Current Study
3. Results
3.1. Clinical Description
3.2. Genetic Testing
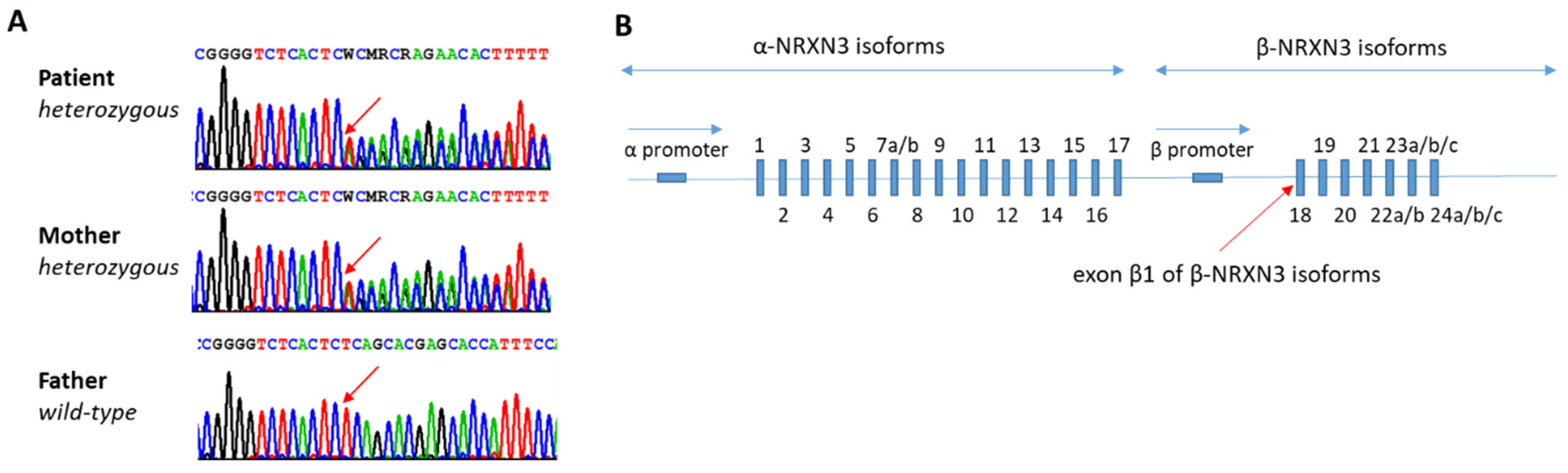
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Geppert, M.; Khvotchev, M.; Krasnoperov, V.; Goda, Y.; Missler, M.; Hammer, R.E.; Ichtchenko, K.; Petrenko, A.G.; Sudhof, T.C. Neurexin Iα is a major alpha-latrotoxin receptor that cooperates in alpha-latrotoxin action. J. Biol. Chem. 1998, 273, 1705–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaags, A.K.; Lionel, A.C.; Sato, D.; Goodenberger, M.; Stein, Q.P.; Curran, S.; Ogilvie, C.; Ahn, J.W.; Drmic, I.; Senman, L.; et al. Rare deletions at the neurexin 3 locus in autism spectrum disorder. Am. J. Hum. Genet. 2012, 90, 133–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Missler, M.; Zhang, W.; Rohlmann, A.; Kattenstroth, G.; Hammer, R.E.; Gottmann, K.; Sudhof, T.C. α-neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature 2003, 423, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.Y.; Chen, L.Y.; Jiang, M.; Trotter, J.H.; Seigneur, E.; Sudhof, T.C. Neurexin-2: An inhibitory neurexin that restricts excitatory synapse formation in the hippocampus. Sci. Adv. 2023, 9, eadd8856. [Google Scholar] [CrossRef] [PubMed]
- Gomez, A.M.; Traunmuller, L.; Scheiffele, P. Neurexins: Molecular codes for shaping neuronal synapses. Nat. Rev. Neurosci. 2021, 22, 137–151. [Google Scholar] [CrossRef]
- Hauser, D.; Behr, K.; Konno, K.; Schreiner, D.; Schmidt, A.; Watanabe, M.; Bischofberger, J.; Scheiffele, P. Targeted proteoform mapping uncovers specific Neurexin-3 variants required for dendritic inhibition. Neuron 2022, 110, 2094–2109.e10. [Google Scholar] [CrossRef]
- Missler, M.; Sudhof, T.C. Neurexins: Three genes and 1001 products. Trends Genet. 1998, 14, 20–26. [Google Scholar] [CrossRef]
- Noborn, F.; Sterky, F.H. Role of neurexin heparan sulfate in the molecular assembly of synapses—Expanding the neurexin code? FEBS J. 2023, 290, 252–265. [Google Scholar] [CrossRef]
- Sudhof, T.C. Neuroligins and neurexins link synaptic function to cognitive disease. Nature 2008, 455, 903–911. [Google Scholar] [CrossRef] [Green Version]
- Tromp, A.; Mowry, B.; Giacomotto, J. Neurexins in autism and schizophrenia-a review of patient mutations, mouse models and potential future directions. Mol. Psychiatry 2021, 26, 747–760. [Google Scholar] [CrossRef]
- Sterky, F.H.; Trotter, J.H.; Lee, S.J.; Recktenwald, C.V.; Du, X.; Zhou, B.; Zhou, P.; Schwenk, J.; Fakler, B.; Sudhof, T.C. Carbonic anhydrase-related protein CA10 is an evolutionarily conserved pan-neurexin ligand. Proc. Natl. Acad. Sci. USA 2017, 114, E1253–E1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treutlein, B.; Gokce, O.; Quake, S.R.; Sudhof, T.C. Cartography of neurexin alternative splicing mapped by single-molecule long-read mRNA sequencing. Proc. Natl. Acad. Sci. USA 2014, 111, E1291–E1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullrich, B.; Ushkaryov, Y.A.; Sudhof, T.C. Cartography of neurexins: More than 1000 isoforms generated by alternative splicing and expressed in distinct subsets of neurons. Neuron 1995, 14, 497–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gresa-Arribas, N.; Planaguma, J.; Petit-Pedrol, M.; Kawachi, I.; Katada, S.; Glaser, C.A.; Simabukuro, M.M.; Armangue, T.; Martinez-Hernandez, E.; Graus, F.; et al. Human neurexin-3alpha antibodies associate with encephalitis and alter synapse development. Neurology 2016, 86, 2235–2242. [Google Scholar] [CrossRef] [Green Version]
- Hansen, N.; Lange, C.; Maass, F.; Hassoun, L.; Bouter, C.; Stocker, W.; Schott, B.H.; Wiltfang, J.; Fitzner, D. Mild Amnestic Cognitive Impairment and Depressive Symptoms in Autoimmune Encephalitis Associated with Serum Anti-Neurexin-3alpha Autoantibodies. Brain Sci. 2021, 11, 673. [Google Scholar] [CrossRef]
- Shiwaku, H.; Katayama, S.; Gao, M.; Kondo, K.; Nakano, Y.; Motokawa, Y.; Toyoda, S.; Yoshida, F.; Hori, H.; Kubota, T.; et al. Analyzing schizophrenia-related phenotypes in mice caused by autoantibodies against NRXN1alpha in schizophrenia. Brain Behav. Immun. 2023, 111, 32–45. [Google Scholar] [CrossRef]
- Waters, P.J.; Irani, S.R. Neurexin-3alpha: A new antibody target in autoimmune encephalitis. Neurology 2016, 86, 2222–2223. [Google Scholar] [CrossRef]
- Zhu, L.; Shang, Q.; Zhao, C.W.; Dai, S.; Wu, Q. Case report: Anti-neurexin-3alpha-associated autoimmune encephalitis secondary to contrast-induced encephalopathy. Front. Neurol. 2023, 14, 1060110. [Google Scholar] [CrossRef]
- Bena, F.; Bruno, D.L.; Eriksson, M.; van Ravenswaaij-Arts, C.; Stark, Z.; Dijkhuizen, T.; Gerkes, E.; Gimelli, S.; Ganesamoorthy, D.; Thuresson, A.C.; et al. Molecular and clinical characterization of 25 individuals with exonic deletions of NRXN1 and comprehensive review of the literature. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2013, 162, 388–403. [Google Scholar] [CrossRef]
- Ching, M.S.; Shen, Y.; Tan, W.H.; Jeste, S.S.; Morrow, E.M.; Chen, X.; Mukaddes, N.M.; Yoo, S.Y.; Hanson, E.; Hundley, R.; et al. Deletions of NRXN1 (neurexin-1) predispose to a wide spectrum of developmental disorders. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2010, 153, 937–947. [Google Scholar] [CrossRef]
- Kirov, G.; Rees, E.; Walters, J.T.; Escott-Price, V.; Georgieva, L.; Richards, A.L.; Chambert, K.D.; Davies, G.; Legge, S.E.; Moran, J.L.; et al. The penetrance of copy number variations for schizophrenia and developmental delay. Biol. Psychiatry 2014, 75, 378–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaaf, C.P.; Boone, P.M.; Sampath, S.; Williams, C.; Bader, P.I.; Mueller, J.M.; Shchelochkov, O.A.; Brown, C.W.; Crawford, H.P.; Phalen, J.A.; et al. Phenotypic spectrum and genotype-phenotype correlations of NRXN1 exon deletions. Eur. J. Hum. Genet. 2012, 20, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Vinas-Jornet, M.; Esteba-Castillo, S.; Gabau, E.; Ribas-Vidal, N.; Baena, N.; San, J.; Ruiz, A.; Coll, M.D.; Novell, R.; Guitart, M. A common cognitive, psychiatric, and dysmorphic phenotype in carriers of NRXN1 deletion. Mol. Genet. Genom. Med. 2014, 2, 512–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curran, S.; Ahn, J.W.; Grayton, H.; Collier, D.A.; Ogilvie, C.M. NRXN1 deletions identified by array comparative genome hybridisation in a clinical case series—Further understanding of the relevance of NRXN1 to neurodevelopmental disorders. J. Mol. Psychiatry 2013, 1, 4. [Google Scholar] [CrossRef] [Green Version]
- Lal, D.; Ruppert, A.K.; Trucks, H.; Schulz, H.; de Kovel, C.G.; Kasteleijn-Nolst Trenite, D.; Sonsma, A.C.; Koeleman, B.P.; Lindhout, D.; Weber, Y.G.; et al. Burden analysis of rare microdeletions suggests a strong impact of neurodevelopmental genes in genetic generalised epilepsies. PLoS Genet. 2015, 11, e1005226. [Google Scholar] [CrossRef]
- Moller, R.S.; Weber, Y.G.; Klitten, L.L.; Trucks, H.; Muhle, H.; Kunz, W.S.; Mefford, H.C.; Franke, A.; Kautza, M.; Wolf, P.; et al. Exon-disrupting deletions of NRXN1 in idiopathic generalized epilepsy. Epilepsia 2013, 54, 256–264. [Google Scholar] [CrossRef]
- Al Shehhi, M.; Forman, E.B.; Fitzgerald, J.E.; McInerney, V.; Krawczyk, J.; Shen, S.; Betts, D.R.; Ardle, L.M.; Gorman, K.M.; King, M.D.; et al. NRXN1 deletion syndrome; phenotypic and penetrance data from 34 families. Eur. J. Med. Genet. 2019, 62, 204–209. [Google Scholar] [CrossRef]
- Gauthier, J.; Siddiqui, T.J.; Huashan, P.; Yokomaku, D.; Hamdan, F.F.; Champagne, N.; Lapointe, M.; Spiegelman, D.; Noreau, A.; Lafreniere, R.G.; et al. Truncating mutations in NRXN2 and NRXN1 in autism spectrum disorders and schizophrenia. Hum. Genet. 2011, 130, 563–573. [Google Scholar] [CrossRef] [Green Version]
- Rochtus, A.M.; Trowbridge, S.; Goldstein, R.D.; Sheidley, B.R.; Prabhu, S.P.; Haynes, R.; Kinney, H.C.; Poduri, A.H. Mutations in NRXN1 and NRXN2 in a patient with early-onset epileptic encephalopathy and respiratory depression. Cold Spring Harb. Mol. Case Stud. 2019, 5, a003442. [Google Scholar] [CrossRef] [Green Version]
- Born, G.; Grayton, H.M.; Langhorst, H.; Dudanova, I.; Rohlmann, A.; Woodward, B.W.; Collier, D.A.; Fernandes, C.; Missler, M. Genetic targeting of NRXN2 in mice unveils role in excitatory cortical synapse function and social behaviors. Front. Synaptic Neurosci. 2015, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Dachtler, J.; Glasper, J.; Cohen, R.N.; Ivorra, J.L.; Swiffen, D.J.; Jackson, A.J.; Harte, M.K.; Rodgers, R.J.; Clapcote, S.J. Deletion of α-neurexin II results in autism-related behaviors in mice. Transl. Psychiatry 2014, 4, e484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Yang, L.; Tang, C.; Liu, K.; Lu, Y.; Wang, H.; Yan, K.; Qiu, Z.; Zhou, W. Mutations of CNTNAP1 led to defects in neuronal development. JCI Insight 2020, 5, e135697. [Google Scholar] [CrossRef] [PubMed]
- Peippo, M.M.; Simola, K.O.; Valanne, L.K.; Larsen, A.T.; Kahkonen, M.; Auranen, M.P.; Ignatius, J. Pitt-Hopkins syndrome in two patients and further definition of the phenotype. Clin. Dysmorphol. 2006, 15, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Zweier, C.; de Jong, E.K.; Zweier, M.; Orrico, A.; Ousager, L.B.; Collins, A.L.; Bijlsma, E.K.; Oortveld, M.A.; Ekici, A.B.; Reis, A.; et al. CNTNAP2 and NRXN1 are mutated in autosomal-recessive Pitt-Hopkins-like mental retardation and determine the level of a common synaptic protein in Drosophila. Am. J. Hum. Genet. 2009, 85, 655–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, W.O. Time-course of cowpea chlorotic mottle virus RNA replication. Intervirology 1978, 9, 119–128. [Google Scholar] [CrossRef]
- Najm, J.; Horn, D.; Wimplinger, I.; Golden, J.A.; Chizhikov, V.V.; Sudi, J.; Christian, S.L.; Ullmann, R.; Kuechler, A.; Haas, C.A.; et al. Mutations of CASK cause an X-linked brain malformation phenotype with microcephaly and hypoplasia of the brainstem and cerebellum. Nat. Genet. 2008, 40, 1065–1067. [Google Scholar] [CrossRef]
- Faheem, M.; Naseer, M.I.; Chaudhary, A.G.; Kumosani, T.A.; Rasool, M.; Algahtani, H.A.; Bibi, F.; Kamal, M.A.; Al-Qahtani, M.H. Array-comparative genomic hybridization analysis of a cohort of Saudi patients with epilepsy. CNS Neurol. Disord. Drug. Targets 2015, 14, 468–475. [Google Scholar] [CrossRef]
- Griswold, A.J.; Ma, D.; Cukier, H.N.; Nations, L.D.; Schmidt, M.A.; Chung, R.H.; Jaworski, J.M.; Salyakina, D.; Konidari, I.; Whitehead, P.L.; et al. Evaluation of copy number variations reveals novel candidate genes in autism spectrum disorder-associated pathways. Hum. Mol. Genet. 2012, 21, 3513–3523. [Google Scholar] [CrossRef] [Green Version]
- Nicita, F.; Di Giacomo, M.; Palumbo, O.; Ferri, E.; Maiorani, D.; Vigevano, F.; Carella, M.; Capuano, A. Neurological features of 14q24–q32 interstitial deletion: Report of a new case. Mol. Cytogenet. 2015, 8, 93. [Google Scholar] [CrossRef] [Green Version]
- Riegel, M.; Moreira, L.M.; Espirito Santo, L.D.; Toralles, M.B.; Schinzel, A. Interstitial 14q24.3 to q31.3 deletion in a 6-year-old boy with a non-specific dysmorphic phenotype. Mol. Cytogenet. 2014, 7, 77. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Wang, Q.; Liu, Y.; Yang, W.; He, Y.; Gusella, J.F.; Song, J.; Shen, Y. A rare exonic NRXN3 deletion segregating with neurodevelopmental and neuropsychiatric conditions in a three-generation Chinese family. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2018, 177, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Rohlmann, A.; Sargsyan, V.; Aramuni, G.; Hammer, R.E.; Sudhof, T.C.; Missler, M. Extracellular domains of α-neurexins participate in regulating synaptic transmission by selectively affecting N- and P/Q-type Ca2+ channels. J. Neurosci. 2005, 25, 4330–4342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, G.R.; Aoto, J.; Tabuchi, K.; Foldy, C.; Covy, J.; Yee, A.X.; Wu, D.; Lee, S.J.; Chen, L.; Malenka, R.C.; et al. β-Neurexins Control Neural Circuits by Regulating Synaptic Endocannabinoid Signaling. Cell 2015, 162, 593–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoto, J.; Foldy, C.; Ilcus, S.M.; Tabuchi, K.; Sudhof, T.C. Distinct circuit-dependent functions of presynaptic neurexin-3 at GABAergic and glutamatergic synapses. Nat. Neurosci. 2015, 18, 997–1007. [Google Scholar] [CrossRef] [Green Version]
- Aoto, J.; Martinelli, D.C.; Malenka, R.C.; Tabuchi, K.; Sudhof, T.C. Presynaptic neurexin-3 alternative splicing trans-synaptically controls postsynaptic AMPA receptor trafficking. Cell 2013, 154, 75–88. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.Y.; Jiang, M.; Zhang, B.; Gokce, O.; Sudhof, T.C. Conditional Deletion of All Neurexins Defines Diversity of Essential Synaptic Organizer Functions for Neurexins. Neuron 2017, 94, 611–625.e4. [Google Scholar] [CrossRef] [Green Version]
- Trotter, J.H.; Wang, C.Y.; Zhou, P.; Nakahara, G.; Sudhof, T.C. A combinatorial code of neurexin-3 alternative splicing controls inhibitory synapses via a trans-synaptic dystroglycan signaling loop. Nat. Commun. 2023, 14, 1771. [Google Scholar] [CrossRef]
- Oku, S.; Siddiqui, T.J. A GPI-anchored Neurexin 3 proteoform mediates dendritic inhibition. Neuron 2022, 110, 2041–2044. [Google Scholar] [CrossRef]
- Khoja, S.; Haile, M.T.; Chen, L.Y. Advances in neurexin studies and the emerging role of neurexin-2 in autism spectrum disorder. Front. Mol. Neurosci. 2023, 16, 1125087. [Google Scholar] [CrossRef]
- Mohrmann, I.; Gillessen-Kaesbach, G.; Siebert, R.; Caliebe, A.; Hellenbroich, Y. A de novo 0.57 Mb microdeletion in chromosome 11q13.1 in a patient with speech problems, autistic traits, dysmorphic features and multiple endocrine neoplasia type 1. Eur. J. Med. Genet. 2011, 54, e461–e464. [Google Scholar] [CrossRef]
- Boyle, M.I.; Jespersgaard, C.; Nazaryan, L.; Ravn, K.; Brondum-Nielsen, K.; Bisgaard, A.M.; Tumer, Z. Deletion of 11q12.3–11q13.1 in a patient with intellectual disability and childhood facial features resembling Cornelia de Lange syndrome. Gene 2015, 572, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.; Berutti, R.; Lorenz-Depiereux, B.; Graf, E.; Eckstein, G.; Mayr, J.A.; Meitinger, T.; Ahting, U.; Prokisch, H.; Strom, T.M.; et al. Mitochondrial DNA mutation analysis from exome sequencing-A more holistic approach in diagnostics of suspected mitochondrial disease. J. Inherit. Metab. Dis. 2019, 42, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Firth, H.V.; Richards, S.M.; Bevan, A.P.; Clayton, S.; Corpas, M.; Rajan, D.; Van Vooren, S.; Moreau, Y.; Pettett, R.M.; Carter, N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 2009, 84, 524–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- UniProt, C. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [Green Version]
- Sobreira, N.; Schiettecatte, F.; Valle, D.; Hamosh, A. GeneMatcher: A matching tool for connecting investigators with an interest in the same gene. Hum. Mutat. 2015, 36, 928–930. [Google Scholar] [CrossRef] [Green Version]
- Lord, C.; Risi, S.; Lambrecht, L.; Cook, E.H., Jr.; Leventhal, B.L.; DiLavore, P.C.; Pickles, A.; Rutter, M. The autism diagnostic observation schedule-generic: A standard measure of social and communication deficits associated with the spectrum of autism. J. Autism Dev. Disord. 2000, 30, 205–223. [Google Scholar] [CrossRef]
- Hishimoto, A.; Liu, Q.R.; Drgon, T.; Pletnikova, O.; Walther, D.; Zhu, X.G.; Troncoso, J.C.; Uhl, G.R. Neurexin 3 polymorphisms are associated with alcohol dependence and altered expression of specific isoforms. Hum. Mol. Genet. 2007, 16, 2880–2891. [Google Scholar] [CrossRef] [PubMed]
- Koolen, D.A.; Morgan, A.; de Vries, B.B.A. Koolen-de Vries Syndrome. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; Springer: Seattle, WA, USA, 1993. [Google Scholar]
- Smith, A.C.M.; Boyd, K.E.; Brennan, C.; Charles, J.; Elsea, S.H.; Finucane, B.M.; Foster, R.; Gropman, A.; Girirajan, S.; Haas-Givler, B. Smith-Magenis Syndrome. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; Springer: Seattle, WA, USA, 1993. [Google Scholar]
- Krgovic, D.; Gorenjak, M.; Rihar, N.; Opalic, I.; Stangler Herodez, S.; Gregoric Kumperscak, H.; Dovc, P.; Kokalj Vokac, N. Impaired Neurodevelopmental Genes in Slovenian Autistic Children Elucidate the Comorbidity of Autism With Other Developmental Disorders. Front. Mol. Neurosci. 2022, 15, 912671. [Google Scholar] [CrossRef] [PubMed]
- Lieberwirth, J.K.; Buttner, B.; Klockner, C.; Platzer, K.; Popp, B.; Abou Jamra, R. AutoCaSc: Prioritizing candidate genes for neurodevelopmental disorders. Hum. Mutat. 2022, 43, 1795–1807. [Google Scholar] [CrossRef] [PubMed]
- Younis, N.S.; Mohamed, M.E.; Alolayan, A.A.; Alhussain, G.Y.; Al-Mousa, H.A.; Alshamrani, J.A.; AlMutayib, M.M.; AlQahtani, M.M.; Alhaddad, Z.A.; Alfarhan, Z.S.; et al. Identification of epilepsy concomitant candidate genes recognized in Saudi epileptic patients. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 2143–2157. [Google Scholar] [CrossRef]
- Kamal, N.; Jafari Khamirani, H.; Dara, M.; Dianatpour, M. NRXN3 mutations cause developmental delay, movement disorder, and behavioral problems: CRISPR edited cells based WES results. Gene 2023, 867, 147347. [Google Scholar] [CrossRef]
- Duong, L.; Klitten, L.L.; Moller, R.S.; Ingason, A.; Jakobsen, K.D.; Skjodt, C.; Didriksen, M.; Hjalgrim, H.; Werge, T.; Tommerup, N. Mutations in NRXN1 in a family multiply affected with brain disorders: NRXN1 mutations and brain disorders. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012, 159, 354–358. [Google Scholar] [CrossRef]
- Harrison, V.; Connell, L.; Hayesmoore, J.; McParland, J.; Pike, M.G.; Blair, E. Compound heterozygous deletion of NRXN1 causing severe developmental delay with early onset epilepsy in two sisters. Am. J. Med. Genet. Part A 2011, 155, 2826–2831. [Google Scholar] [CrossRef]
- Falivelli, G.; De Jaco, A.; Favaloro, F.L.; Kim, H.; Wilson, J.; Dubi, N.; Ellisman, M.H.; Abrahams, B.S.; Taylor, P.; Comoletti, D. Inherited genetic variants in autism-related CNTNAP2 show perturbed trafficking and ATF6 activation. Hum. Mol. Genet. 2012, 21, 4761–4773. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feichtinger, R.G.; Preisel, M.; Brugger, K.; Wortmann, S.B.; Mayr, J.A. Case Report—An Inherited Loss-of-Function NRXN3 Variant Potentially Causes a Neurodevelopmental Disorder with Autism Consistent with Previously Described 14q24.3-31.1 Deletions. Genes 2023, 14, 1217. https://doi.org/10.3390/genes14061217
Feichtinger RG, Preisel M, Brugger K, Wortmann SB, Mayr JA. Case Report—An Inherited Loss-of-Function NRXN3 Variant Potentially Causes a Neurodevelopmental Disorder with Autism Consistent with Previously Described 14q24.3-31.1 Deletions. Genes. 2023; 14(6):1217. https://doi.org/10.3390/genes14061217
Chicago/Turabian StyleFeichtinger, René G., Martin Preisel, Karin Brugger, Saskia B. Wortmann, and Johannes A. Mayr. 2023. "Case Report—An Inherited Loss-of-Function NRXN3 Variant Potentially Causes a Neurodevelopmental Disorder with Autism Consistent with Previously Described 14q24.3-31.1 Deletions" Genes 14, no. 6: 1217. https://doi.org/10.3390/genes14061217
APA StyleFeichtinger, R. G., Preisel, M., Brugger, K., Wortmann, S. B., & Mayr, J. A. (2023). Case Report—An Inherited Loss-of-Function NRXN3 Variant Potentially Causes a Neurodevelopmental Disorder with Autism Consistent with Previously Described 14q24.3-31.1 Deletions. Genes, 14(6), 1217. https://doi.org/10.3390/genes14061217