
Inherited Reticulate Pigmentary Disorders



Abstract
:1. Introduction
2. Search Strategy
3. Dyschromatosis Symmetrica Hereditaria
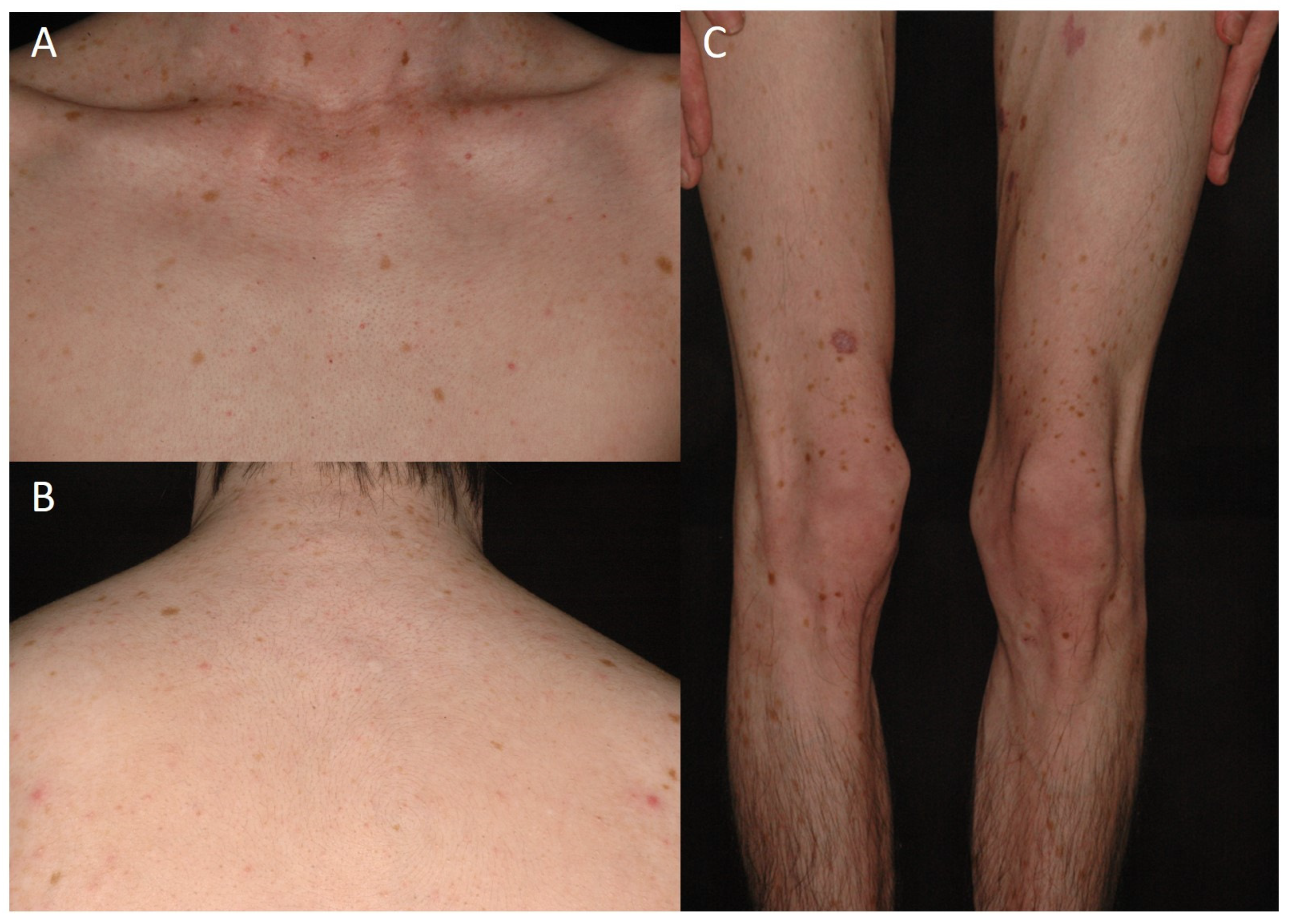
3.1. Clinical and Histological Feature
3.2. Genetic Architecture
4. Dyschromatosis Universalis Hereditaria
4.1. Clinical and Histological Feature
4.2. Genetic Architecture
5. Reticulate Acropigmentation of Kitamura
5.1. Clinical and Histological Feature
5.2. Genetic Architecture
6. Dowling-Degos Disease
6.1. Clinical and Histological Feature
6.2. Genetic Architecture
7. Dyskeratosis Congenita
7.1. Clinical and Histological Feature
7.2. Genetic Architecture
8. Naegeli–Franceschetti–Jadassohn Syndrome
8.1. Clinical and Histological Feature
8.2. Genetic Architecture
9. Dermatopathia Pigmentosa Reticularis
9.1. Clinical and Histological Feature
9.2. Genetic Architecture
10. X-Linked Reticulate Pigmentary Disorder
10.1. Clinical and Histological Feature
10.2. Genetic Architecture
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, J.; Li, M.; Yao, Z. Updated review of genetic reticulate pigmentary disorders. Br. J. Dermatol. 2017, 177, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Sardana, K.; Goel, K.; Chugh, S. Reticulate pigmentary disorders. Indian. J. Dermatol. Venereol. Leprol. 2013, 79, 17–29. [Google Scholar] [CrossRef]
- Toyama, I. Dyschromatosis symmetrica hereditarian. Jpn. J. Dermatol. Urol. 1929, 27, 95–96. [Google Scholar]
- Sheu, H.M.; Yu, H.S. Dyschromatosis symmetrica hereditaria—A histochemical and ultrastructural study. Taiwan Yi Xue Hui Za Zhi 1985, 84, 238–249. [Google Scholar]
- Chao, S.-C.; Lee, J.Y.-Y.; Sheu, H.-M.; Yang, M.-H. A novel deletion mutation of the DSRAD gene in a Taiwanese patient with dyschromatosis symmetrica hereditaria. Br. J. Dermatol. 2005, 153, 1064–1066. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Chao, S.-C.; Yang, M.-H.; Yang, C.-C. A novel mutation of the RNA-specific adenosine deaminase 1 gene in a Taiwanese patient with dyschromatosis symmetrica hereditaria and Becker’s nevus-like lesion. Dermatol. Sin. 2016, 34, 110–111. [Google Scholar] [CrossRef] [Green Version]
- Peng, A.C.-Y.; Chen, Y.-A.; Chao, S.-C. Dyschromatosis symmetrica hereditaria: A retrospective case series and literature review. Dermatol. Sin. 2013, 31, 19–24. [Google Scholar] [CrossRef]
- Hayashi, M.; Suzuki, T. Dyschromatosis symmetrica hereditaria. J. Dermatol. 2013, 40, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-A.; Chao, S.-C.; Lee, J. A novel deletion mutation in the adenosine deaminase RNA-specific gene in a Taiwanese patient with dyschromatosis symmetrica hereditaria. Dermatol. Sin. 2011, 29, 109–110. [Google Scholar] [CrossRef] [Green Version]
- Ostlere, L.S.; Ratnavel, R.C.; Lawlor, F.; Black, M.M.; Griffiths, W.A. Reticulate acropigmentation of Dohi. Clin. Exp. Dermatol. 1995, 20, 477–479. [Google Scholar] [CrossRef] [PubMed]
- Oyama, M.; Shimizu, H.; Ohata, Y.; Tajima, S.; Nishikawa, T. Dyschromatosis symmetrica hereditaria (reticulate acropigmentation of Dohi): Report of a Japanese family with the condition and a literature review of 185 cases. Br. J. Dermatol. 1999, 140, 491–496. [Google Scholar] [CrossRef]
- He, P.P.; He, C.D.; Cui, Y.; Yang, S.; Xu, H.H.; Li, M.; Yuan, W.T.; Gao, M.; Liang, Y.H.; Li, C.R.; et al. Refined localization of dyschromatosis symmetrica hereditaria gene to a 9.4-cM region at 1q21-22 and a literature review of 136 cases reported in China. Br. J. Dermatol. 2004, 150, 633–639. [Google Scholar] [CrossRef]
- Urabe, K.; Hori, Y. Dyschromatosis. Semin. Cutan. Med. Surg. 1997, 16, 81–85. [Google Scholar] [CrossRef]
- Kondo, T.; Suzuki, T.; Ito, S.; Kono, M.; Negoro, T.; Tomita, Y. Dyschromatosis symmetrica hereditaria associated with neurological disorders. J. Dermatol. 2008, 35, 662–666. [Google Scholar] [CrossRef]
- Miyamura, Y.; Suzuki, T.; Kono, M.; Inagaki, K.; Ito, S.; Suzuki, N.; Tomita, Y. Mutations of the RNA-specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria. Am. J. Hum. Genet. 2003, 73, 693–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kono, M.; Sugiura, K.; Suganuma, M.; Hayashi, M.; Takama, H.; Suzuki, T.; Matsunaga, K.; Tomita, Y.; Akiyama, M. Whole-exome sequencing identifies ADAM10 mutations as a cause of reticulate acropigmentation of Kitamura, a clinical entity distinct from Dowling-Degos disease. Hum. Mol. Genet. 2013, 22, 3524–3533. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zeng, Y.; Murray, J.M.; Nishikura, K. Genomic organization and chromosomal location of the human dsRNA adenosine deaminase gene: The enzyme for glutamate-activated ion channel RNA editing. J. Mol. Biol. 1995, 254, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.J.; Gao, M.; Li, M.; Li, M.; Li, C.R.; Cui, Y.; He, P.P.; Xu, S.J.; Xiong, X.Y.; Wang, Z.X.; et al. Identification of a locus for dyschromatosis symmetrica hereditaria at chromosome 1q11-1q21. J. Investig. Dermatol. 2003, 120, 776–780. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Liu, W.; Jiang, L.; Sun, M.; Ao, Y.; Zhao, X.; Song, Y.; Luo, Y.; Lo, W.H.; Zhang, X. Novel mutations of the RNA-specific adenosine deaminase gene (DSRAD) in Chinese families with dyschromatosis symmetrica hereditaria. J. Investig. Dermatol. 2004, 122, 896–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bass, B.L.; Weintraub, H. An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell 1988, 55, 1089–1098. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, L.; Huang, P.; Xiong, J.; Xiao, Y.; Wang, C.; Mao, D.; Liu, L. Case Report: Aicardi-Goutières Syndrome Type 6 and Dyschromatosis Symmetrica Hereditaria With Congenital Heart Disease and Mitral Valve Calcification—Phenotypic Variants Caused by Adenosine Deaminase Acting on the RNA 1 Gene Homozygous Mutations. Front. Pediatr. 2022, 10, 852903. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhang, L.; Zhang, Y.; Luo, H.; Zhu, L.; Wang, P.; Zhang, G.; Wang, X. Two novel ADAR1 gene mutations in two patients with dyschromatosis symmetrical hereditaria from birth. Mol. Med. Rep. 2017, 15, 3715–3718. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, N.; Suzuki, T.; Inagaki, K.; Ito, S.; Kono, M.; Fukai, K.; Takama, H.; Sato, K.; Ishikawa, O.; Abe, M.; et al. Mutation analysis of the ADAR1 gene in dyschromatosis symmetrica hereditaria and genetic differentiation from both dyschromatosis universalis hereditaria and acropigmentatio reticularis. J. Investig. Dermatol. 2005, 124, 1186–1192. [Google Scholar] [CrossRef] [Green Version]
- Chin, Y.-Y.; Chen, G.-S.; Hu, S.C.-S.; Lan, C.-C.E. Dyschromatosis universalis hereditaria: A familial case with ultrastructural skin investigation. Dermatologica Sinica 2011, 29, 137–141. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Li, D.; Zhang, J.; Chen, X.; Huang, M.; Archacki, S.; Tian, Y.; Ren, W.; Mei, A.; Zhang, Q.; et al. Mutations in ABCB6 cause dyschromatosis universalis hereditaria. J. Investig. Dermatol. 2013, 133, 2221–2228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Hawsawi, K.; Al Aboud, K.; Ramesh, V.; Al Aboud, D. Dyschromatosis universalis hereditaria: Report of a case and review of the literature. Pediatr. Dermatol. 2002, 19, 523–526. [Google Scholar] [CrossRef]
- Yadalla, H.K.; Pinninti, S.; Babu, A.R. Dyschromatosis universalis hereditaria: Infrequent genodermatoses in India. Indian. J. Hum. Genet. 2013, 19, 487–490. [Google Scholar] [CrossRef] [Green Version]
- Sorensen, R.H.; Werner, K.A.; Kobayashi, T.T. Dyschromatosis Universalis Hereditaria with Oral Leukokeratosis—A Case of Mistaken Identity and Review of the Literature. Pediatr. Dermatol. 2015, 32, e283–e287. [Google Scholar] [CrossRef]
- Cao, L.; Zhang, R.; Yong, L.; Chen, S.; Zhang, H.; Chen, W.; Xu, Q.; Ge, H.; Mao, Y.; Zhen, Q.; et al. Novel missense mutation of SASH1 in a Chinese family with dyschromatosis universalis hereditaria. BMC Med. Genom. 2021, 14, 168. [Google Scholar] [CrossRef]
- Zhou, D.; Wei, Z.; Deng, S.; Wang, T.; Zai, M.; Wang, H.; Guo, L.; Zhang, J.; Zhong, H.; He, L.; et al. SASH1 regulates melanocyte transepithelial migration through a novel Galphas-SASH1-IQGAP1-E-Cadherin dependent pathway. Cell Signal. 2013, 25, 1526–1538. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Tang, L.; Li, X.; Dai, Y.; Zheng, X.; Gao, M.; Wang, P. Identification of a Novel Mutation in SASH1 Gene in a Chinese Family With Dyschromatosis Universalis Hereditaria and Genotype-Phenotype Correlation Analysis. Front. Genet. 2020, 11, 841. [Google Scholar] [CrossRef]
- Liu, J.W.; Jun Sun, A.; Vano-Galvan, S.; Liu, F.X.; Wei, X.X.; Ma, D.L. Differential Diagnosis of Two Chinese Families with Dyschromatoses by Targeted Gene Sequencing. Chin. Med. J. 2016, 129, 33–38. [Google Scholar] [CrossRef]
- Liu, J.W.; Habulieti, X.; Wang, R.R.; Ma, D.L.; Zhang, X. Two novel SASH1 mutations in Chinese families with dyschromatosis universalis hereditaria. J. Clin. Lab. Anal. 2021, 35, e23803. [Google Scholar] [CrossRef]
- Stuhrmann, M.; Hennies, H.C.; Bukhari, I.A.; Brakensiek, K.; Nurnberg, G.; Becker, C.; Huebener, J.; Miranda, M.C.; Frye-Boukhriss, H.; Knothe, S.; et al. Dyschromatosis universalis hereditaria: Evidence for autosomal recessive inheritance and identification of a new locus on chromosome 12q21-q23. Clin. Genet. 2008, 73, 566–572. [Google Scholar] [CrossRef]
- Kitamura, K.; Akamatsu, S.; Hirokawa, K. A special form of acropigmentation: Acropigmentation reticularis. Hautarzt 1953, 4, 152–156. [Google Scholar]
- Kono, M.; Akiyama, M. Dyschromatosis symmetrica hereditaria and reticulate acropigmentation of Kitamura: An update. J. Dermatol. Sci. 2019, 93, 75–81. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, W.A. Reticulate acropigmentation of Kitamura. Br. J. Dermatol. 1976, 95, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Adya, K.A.; Inamadar, A.C.; Palit, A. Reticulate Acropigmentation of Kitamura: A Dermoscopic Perspective. Indian. Dermatol. Online J. 2020, 11, 128–130. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Zhong, W.; Wang, H.; Yang, Y.; Lin, Z. Reticulate acropigmentation of Kitamura with a novel mutation in ADAM10. Clin. Exp. Dermatol. 2019, 44, 700–703. [Google Scholar] [CrossRef] [PubMed]
- Tharmarajah, G.; Faas, L.; Reiss, K.; Saftig, P.; Young, A.; Van Raamsdonk, C.D. Adam10 haploinsufficiency causes freckle-like macules in Hairless mice. Pigment. Cell Melanoma Res. 2012, 25, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Alshaikh, H.; Alsaif, F.; Aldukhi, S. Clinical and Genetic Review of Hereditary Acral Reticulate Pigmentary Disorders. Dermatol. Res. Pract. 2017, 2017, 3518568. [Google Scholar] [CrossRef] [Green Version]
- Kono, M.; Sawada, M.; Nakazawa, Y.; Ogi, T.; Muro, Y.; Akiyama, M. A Japanese Case of Galli-Galli Disease due to a Previously Unreported POGLUT1 Mutation. Acta Derm. Venereol. 2019, 99, 458–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arundhathi, S.; Rajagopal, P.; Gopinath, H.; Rupa Ramani, J. Follicular Dowling-Degos Disease Camouflaged as Comedones: A Case Report and Literature Review. Cureus 2022, 14, e26078. [Google Scholar] [CrossRef]
- El Shabrawi-Caelen, L.; Rütten, A.; Kerl, H. The expanding spectrum of Galli-Galli disease. J. Am. Acad. Dermatol. 2007, 56, S86–S91. [Google Scholar] [CrossRef]
- Gilchrist, H.; Jackson, S.; Morse, L.; Nicotri, T.; Nesbitt, L.T. Galli-Galli disease: A case report with review of the literature. J. Am. Acad. Dermatol. 2008, 58, 299–302. [Google Scholar] [CrossRef]
- Wu, Y.H.; Lin, Y.C. Generalized Dowling-Degos disease. J. Am. Acad. Dermatol. 2007, 57, 327–334. [Google Scholar] [CrossRef]
- Yu, W.-T.; Su, Y.-S.; Lee, C.-H. A Taiwanese woman with Dowling-Degos disease: An electron microscopic study with pathophysiological significance. Dermatologica Sinica 2014, 32, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Stephan, C.; Kurban, M.; Abbas, O. Dowling-Degos disease: A review. Int. J. Dermatol. 2021, 60, 944–950. [Google Scholar] [CrossRef]
- Betz, R.C.; Planko, L.; Eigelshoven, S.; Hanneken, S.; Pasternack, S.M.; Bussow, H.; Van Den Bogaert, K.; Wenzel, J.; Braun-Falco, M.; Rutten, A.; et al. Loss-of-function mutations in the keratin 5 gene lead to Dowling-Degos disease. Am. J. Hum. Genet. 2006, 78, 510–519. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Cheng, R.; Liang, J.; Yan, H.; Zhang, H.; Yang, L.; Li, C.; Jiao, Q.; Lu, Z.; He, J.; et al. Mutations in POFUT1, encoding protein O-fucosyltransferase 1, cause generalized Dowling-Degos disease. Am. J. Hum. Genet. 2013, 92, 895–903. [Google Scholar] [CrossRef] [Green Version]
- Li, C.R.; Xing, Q.H.; Li, M.; Qin, W.; Yue, X.Z.; Zhang, X.J.; Ma, H.J.; Wang, D.G.; Feng, G.Y.; Zhu, W.Y.; et al. A gene locus responsible for reticulate pigmented anomaly of the flexures maps to chromosome 17p13.3. J. Investig. Dermatol. 2006, 126, 1297–1301. [Google Scholar] [CrossRef] [PubMed]
- Basmanav, F.B.; Oprisoreanu, A.M.; Pasternack, S.M.; Thiele, H.; Fritz, G.; Wenzel, J.; Grosser, L.; Wehner, M.; Wolf, S.; Fagerberg, C.; et al. Mutations in POGLUT1, encoding protein O-glucosyltransferase 1, cause autosomal-dominant Dowling-Degos disease. Am. J. Hum. Genet. 2014, 94, 135–143. [Google Scholar] [CrossRef] [Green Version]
- Agut-Busquet, E.; González-Villanueva, I.; Romani de Gabriel, J.; Pascual, J.C.; Ribera Pibernat, M.; Luelmo, J. Dowling-Degos Disease and Hidradenitis Suppurativa. Epidemiological and Clinical Study of 15 Patients and Review of the Literature. Acta Derm. Venereol. 2019, 99, 917–918. [Google Scholar] [CrossRef] [Green Version]
- Pace, N.P.; Mintoff, D.; Borg, I. The Genomic Architecture of Hidradenitis Suppurativa-A Systematic Review. Front. Genet. 2022, 13, 861241. [Google Scholar] [CrossRef] [PubMed]
- Ralser, D.J.; Basmanav, F.B.; Tafazzoli, A.; Wititsuwannakul, J.; Delker, S.; Danda, S.; Thiele, H.; Wolf, S.; Busch, M.; Pulimood, S.A.; et al. Mutations in gamma-secretase subunit-encoding PSENEN underlie Dowling-Degos disease associated with acne inversa. J. Clin. Investig. 2017, 127, 1485–1490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.; Wu, X.; Zhou, Y.; Sheng, L.; Jena, P.K.; Han, D.; Wan, Y.J.Y.; Hwang, S.T. A Western Diet, but Not a High-Fat and Low-Sugar Diet, Predisposes Mice to Enhanced Susceptibility to Imiquimod-Induced Psoriasiform Dermatitis. J. Investig. Dermatol. 2019, 139, 1404–1407. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Wu, X.; Shi, Z.; Huynh, M.; Jena, P.K.; Sheng, L.; Zhou, Y.; Han, D.; Wan, Y.Y.; Hwang, S.T. Diet-induced obesity exacerbates imiquimod-mediated psoriasiform dermatitis in anti-PD-1 antibody-treated mice: Implications for patients being treated with checkpoint inhibitors for cancer. J. Dermatol. Sci. 2020, 97, 194–200. [Google Scholar] [CrossRef]
- Yu, S.; Lee, C.W.; Li, Y.A.; Chen, T.H.; Yu, H.S. Prenatal infection predisposes offspring to enhanced susceptibility to imiquimod-mediated psoriasiform dermatitis in mice. Dermatol. Sin. 2022, 40, 14–19. [Google Scholar] [CrossRef]
- Zhou, S.; Yao, Z. Roles of Infection in Psoriasis. Int. J. Mol. Sci. 2022, 23, 6955. [Google Scholar] [CrossRef]
- Savage, S.A. Human telomeres and telomere biology disorders. Prog. Mol. Biol. Transl. Sci. 2014, 125, 41–66. [Google Scholar] [CrossRef]
- AlSabbagh, M.M. Dyskeratosis congenita: A literature review. J. Dtsch. Dermatol. Ges. 2020, 18, 943–967. [Google Scholar] [CrossRef] [PubMed]
- Niewisch, M.R.; Savage, S.A. An update on the biology and management of dyskeratosis congenita and related telomere biology disorders. Expert. Rev. Hematol. 2019, 12, 1037–1052. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Kumar, A. Dyskeratosis congenita. Adv. Exp. Med. Biol. 2010, 685, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Dokal, I. Dyskeratosis congenita. Hematology Am. Soc. Hematol. Educ. Program. 2011, 2011, 480–486. [Google Scholar] [CrossRef] [Green Version]
- Vulliamy, T.; Dokal, I. Dyskeratosis congenita. Semin. Hematol. 2006, 43, 157–166. [Google Scholar] [CrossRef]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT promoter mutations in familial and sporadic melanoma. Science 2013, 339, 959–961. [Google Scholar] [CrossRef] [Green Version]
- Savage, S.A.; Giri, N.; Baerlocher, G.M.; Orr, N.; Lansdorp, P.M.; Alter, B.P. TINF2, a component of the shelterin telomere protection complex, is mutated in dyskeratosis congenita. Am. J. Hum. Genet. 2008, 82, 501–509. [Google Scholar] [CrossRef] [Green Version]
- Ballew, B.J.; Yeager, M.; Jacobs, K.; Giri, N.; Boland, J.; Burdett, L.; Alter, B.P.; Savage, S.A. Germline mutations of regulator of telomere elongation helicase 1, RTEL1, in Dyskeratosis congenita. Hum. Genet. 2013, 132, 473–480. [Google Scholar] [CrossRef] [Green Version]
- Amin Guldmann, S.; Byrjalsen, A.; Shaker, S.; Elberling, J. A New Pathogenic Variant of the RTEL1 Gene and Dyskeratosis Congenita: A Dermatological View. Acta Derm. Venereol. 2022, 102, adv00710. [Google Scholar] [CrossRef]
- Revesz, T.; Fletcher, S.; al-Gazali, L.I.; DeBuse, P. Bilateral retinopathy, aplastic anaemia, and central nervous system abnormalities: A new syndrome? J. Med. Genet. 1992, 29, 673–675. [Google Scholar] [CrossRef]
- Tummala, H.; Collopy, L.C.; Walne, A.J.; Ellison, A.; Cardoso, S.; Aksu, T.; Yarali, N.; Aslan, D.; Fikret Akata, R.; Teo, J.; et al. Homozygous OB-fold variants in telomere protein TPP1 are associated with dyskeratosis congenita-like phenotypes. Blood 2018, 132, 1349–1353. [Google Scholar] [CrossRef]
- Walne, A.J.; Vulliamy, T.; Marrone, A.; Beswick, R.; Kirwan, M.; Masunari, Y.; Al-Qurashi, F.H.; Aljurf, M.; Dokal, I. Genetic heterogeneity in autosomal recessive dyskeratosis congenita with one subtype due to mutations in the telomerase-associated protein NOP10. Hum. Mol. Genet. 2007, 16, 1619–1629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, U.T. How a single protein complex accommodates many different H/ACA RNAs. Trends Biochem. Sci. 2006, 31, 311–315. [Google Scholar] [CrossRef] [Green Version]
- Shao, Y.; Feng, S.; Huang, J.; Huo, J.; You, Y.; Zheng, Y. A unique homozygous WRAP53 Arg298Trp mutation underlies dyskeratosis congenita in a Chinese Han family. BMC Med. Genet. 2018, 19, 40. [Google Scholar] [CrossRef]
- Marrone, A.; Walne, A.; Tamary, H.; Masunari, Y.; Kirwan, M.; Beswick, R.; Vulliamy, T.; Dokal, I. Telomerase reverse-transcriptase homozygous mutations in autosomal recessive dyskeratosis congenita and Hoyeraal-Hreidarsson syndrome. Blood 2007, 110, 4198–4205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tummala, H.; Walne, A.; Collopy, L.; Cardoso, S.; de la Fuente, J.; Lawson, S.; Powell, J.; Cooper, N.; Foster, A.; Mohammed, S.; et al. Poly(A)-specific ribonuclease deficiency impacts telomere biology and causes dyskeratosis congenita. J. Clin. Investig. 2015, 125, 2151–2160. [Google Scholar] [CrossRef]
- Kocak, H.; Ballew, B.J.; Bisht, K.; Eggebeen, R.; Hicks, B.D.; Suman, S.; O’Neil, A.; Giri, N.; Laboratory, N.D.C.G.R.; Group, N.D.C.S.W.; et al. Hoyeraal-Hreidarsson syndrome caused by a germline mutation in the TEL patch of the telomere protein TPP1. Genes. Dev. 2014, 28, 2090–2102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touzot, F.; Callebaut, I.; Soulier, J.; Gaillard, L.; Azerrad, C.; Durandy, A.; Fischer, A.; de Villartay, J.P.; Revy, P. Function of Apollo (SNM1B) at telomere highlighted by a splice variant identified in a patient with Hoyeraal-Hreidarsson syndrome. Proc. Natl. Acad. Sci. USA 2010, 107, 10097–10102. [Google Scholar] [CrossRef] [Green Version]
- Kermasson, L.; Churikov, D.; Awad, A.; Smoom, R.; Lainey, E.; Touzot, F.; Audebert-Bellanger, S.; Haro, S.; Roger, L.; Costa, E.; et al. Inherited human Apollo deficiency causes severe bone marrow failure and developmental defects. Blood 2022, 139, 2427–2440. [Google Scholar] [CrossRef]
- Sparrow, G.P.; Samman, P.D.; Wells, R.S. Hyperpigmentation and hypohidrosis. (The Naegeli-Franceschetti-Jadassohn syndrome): Report of a family and review of the literature. Clin. Exp. Dermatol. 1976, 1, 127–140. [Google Scholar] [CrossRef]
- Tubaigy, S.M.; Hassan, H.M. Naegeli-Franceschetti-Jadassohn syndrome in a Saudi Arabian family. J. Forensic Sci. 2014, 59, 555–558. [Google Scholar] [CrossRef] [PubMed]
- Heimer, W.L., 2nd; Brauner, G.; James, W.D. Dermatopathia pigmentosa reticularis: A report of a family demonstrating autosomal dominant inheritance. J. Am. Acad. Dermatol. 1992, 26, 298–301. [Google Scholar] [CrossRef]
- Shah, B.J.; Jagati, A.K.; Gupta, N.P.; Dhamale, S.S. Naegeli-Franceschetti-Jadassohn syndrome: A rare case. Indian Dermatol. Online J. 2015, 6, 403–406. [Google Scholar] [CrossRef]
- Burger, B.; Spoerri, I.; Imahorn, E.; Wariwoda, H.; Leeb, T.; Itin, P.H. Naegeli-Franceschetti-Jadassohn syndrome and dermatopathia pigmentosa reticularis: Intrafamilial overlap of phenotypes in patients with the same KRT14 frameshift variant. Br. J. Dermatol. 2019, 181, 864–866. [Google Scholar] [CrossRef]
- Sprecher, E.; Itin, P.; Whittock, N.V.; McGrath, J.A.; Meyer, R.; DiGiovanna, J.J.; Bale, S.J.; Uitto, J.; Richard, G. Refined mapping of Naegeli-Franceschetti-Jadassohn syndrome to a 6 cM interval on chromosome 17q11.2-q21 and investigation of candidate genes. J. Investig. Dermatol. 2002, 119, 692–698. [Google Scholar] [CrossRef]
- Ralser, D.J.; Kumar, S.; Borisov, O.; Sarig, O.; Richard, G.; Wolf, S.; Krawitz, P.M.; Sprecher, E.; Kreiß, M.; Betz, R.C. Identification of a founder mutation in KRT14 associated with Naegeli-Franceschetti-Jadassohn syndrome. Br. J. Dermatol. 2020, 183, 756–757. [Google Scholar] [CrossRef] [PubMed]
- Lugassy, J.; Itin, P.; Ishida-Yamamoto, A.; Holland, K.; Huson, S.; Geiger, D.; Hennies, H.C.; Indelman, M.; Bercovich, D.; Uitto, J.; et al. Naegeli-Franceschetti-Jadassohn syndrome and dermatopathia pigmentosa reticularis: Two allelic ectodermal dysplasias caused by dominant mutations in KRT14. Am. J. Hum. Genet. 2006, 79, 724–730. [Google Scholar] [CrossRef] [Green Version]
- Brar, B.K.; Mehta, V.; Kubba, A. Dermatopathia pigmentosa reticularis. Pediatr. Dermatol. 2007, 24, 566–570. [Google Scholar] [CrossRef]
- Bu, T.S.; Kim, Y.K.; Whang, K.U. A case of dermatopathia pigmentosa reticularis. J. Dermatol. 1997, 24, 266–269. [Google Scholar] [CrossRef]
- Al-Hamdi, K.I.; Ismael, D.K.; Qais Saadoon, A. Dermatopathia pigmentosa reticularis: A report of a case with delayed onset alopecia and onychodystrophy. JAAD Case Rep. 2019, 5, 379–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer-Mueller, C.; Osborn, M.J.; Tolar, J.; Boull, C.; Ebens, C.L. Revertant Mosaicism in Epidermolysis Bullosa. Biomedicines 2022, 10, 114. [Google Scholar] [CrossRef]
- Smith, F.J.; Morley, S.M.; McLean, W.H. Novel mechanism of revertant mosaicism in Dowling-Meara epidermolysis bullosa simplex. J. Investig. Dermatol. 2004, 122, 73–77. [Google Scholar] [CrossRef] [Green Version]
- Schuilenga-Hut, P.H.; Scheffer, H.; Pas, H.H.; Nijenhuis, M.; Buys, C.H.; Jonkman, M.F. Partial revertant mosaicism of keratin 14 in a patient with recessive epidermolysis bullosa simplex. J. Investig. Dermatol. 2002, 118, 626–630. [Google Scholar] [CrossRef] [Green Version]
- van den Akker, P.C.; Bolling, M.C.; Pasmooij, A.M.G. Revertant Mosaicism in Genodermatoses: Natural Gene Therapy Right before Your Eyes. Biomedicines 2022, 10, 2118. [Google Scholar] [CrossRef]
- Partington, M.W.; Marriott, P.J.; Prentice, R.S.; Cavaglia, A.; Simpson, N.E. Familial cutaneous amyloidosis with systemic manifestations in males. Am. J. Med. Genet. 1981, 10, 65–75. [Google Scholar] [CrossRef]
- Pezzani, L.; Brena, M.; Callea, M.; Colombi, M.; Tadini, G. X-linked reticulate pigmentary disorder with systemic manifestations: A new family and review of the literature. Am. J. Med. Genet. A 2013, 161A, 1414–1420. [Google Scholar] [CrossRef]
- Zhao, Y.K.; Fan, L.H.; Lu, J.F.; Luo, Z.Y.; Lin, Z.M.; Wang, H.J.; Luo, D.Q. X-linked reticulate pigmentary disorder in a 4-year-old boy. Postepy Dermatol. Alergol. 2022, 39, 410–412. [Google Scholar] [CrossRef]
- Kim, B.S.; Seo, S.H.; Jung, H.D.; Kwon, K.S.; Kim, M.B. X-Linked reticulate pigmentary disorder in a female patient. Int. J. Dermatol. 2010, 49, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Yu, H.S. A patient with subacute cutaneous lupus erythematosus along Blaschko lines: Implications for the role of keratinocytes in lupus erythematosus. Dermatol. Sin. 2016, 34, 144–147. [Google Scholar] [CrossRef] [Green Version]
- Anderson, R.C.; Zinn, A.R.; Kim, J.; Carder, K.R. X-linked reticulate pigmentary disorder with systemic manifestations: Report of a third family and literature review. Pediatr. Dermatol. 2005, 22, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Ades, L.C.; Rogers, M.; Sillence, D.O. An X-linked reticulate pigmentary disorder with systemic manifestations: Report of a second family. Pediatr. Dermatol. 1993, 10, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Megarbane, H.; Boehm, N.; Chouery, E.; Bernard, R.; Salem, N.; Halaby, E.; Levy, N.; Megarbane, A. X-linked reticulate pigmentary layer. Report of a new patient and demonstration of a skewed X-inactivation. Genet. Couns. 2005, 16, 85–89. [Google Scholar] [PubMed]
- Starokadomskyy, P.; Gemelli, T.; Rios, J.J.; Xing, C.; Wang, R.C.; Li, H.; Pokatayev, V.; Dozmorov, I.; Khan, S.; Miyata, N.; et al. DNA polymerase-alpha regulates the activation of type I interferons through cytosolic RNA:DNA synthesis. Nat. Immunol. 2016, 17, 495–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starokadomskyy, P.; Sifuentes-Dominguez, L.; Gemelli, T.; Zinn, A.R.; Dossi, M.T.; Mellado, C.; Bertrand, P.; Borzutzky, A.; Burstein, E. Evolution of the skin manifestations of X-linked pigmentary reticulate disorder. Br. J. Dermatol. 2017, 177, e200–e201. [Google Scholar] [CrossRef] [PubMed]
- Legeret, C.; Meyer, B.J.; Rovina, A.; Deigendesch, N.; Berger, C.T.; Daikeler, T.; Heijnen, I.; Burstein, E.; Kohler, H.; Recher, M. JAK Inhibition in a Patient with X-Linked Reticulate Pigmentary Disorder. J. Clin. Immunol. 2021, 41, 212–216. [Google Scholar] [CrossRef]

{kind=link}
| Disease | Responsible Gene | Prevalent Ethnicity | Pigmentation Pattern | Other Clinical Manifestations |
|---|---|---|---|---|
| Dyschromatosis symmmetrica hereditarian | ADAR1 | East Asian | mottled hypopigmented and hyperpigmented macules over the dorsal aspects of the extremities | congenital heart disease, hemangioma disease, neurological symptoms |
| Dyschromatosis universalis hereditaria | SASH1 for DUH1, chromosome 12q21-q23 for DUH2, ABCB6 for DUH3 | East Asian | mottled hyperpigmented and hypopigmented macules of irregular size and shape distributed randomly all over the body | abnormalities of hair and nails |
| Reticulate acropigmentation of Kitamura | ADAM10 | East Asian | angular reticulate, freckle-like hyperpigmented macules distributed on the dorsal aspect of the extremities | epidermal atrophy |
| Dowling-Degos disease | KRT5 for DDD1, POFUT1 for DDD2, chromosome 17p13.3 for DDD3, POGLUT1 for DDD4 | Caucasian | reticulate, dot-like hyperpigmentation of flexures | comedo-like follicular papules |
| Dyskeratosis congenita | DKC1 for DKCX, TERT for DKCA2, TINF2 for DKCA3, RTEL1 for DKCA4, TINF2 for DKCA5, ACD for DKCA6, NOP10 for DKCB1, NHP2 for DKCB2, WRAP53 for DKCB3, TERT for DKCB4, RTEL1 for DKCB5, PARN for DKCB6, ACD for DKCB7, DCLRE1B for DKCB8 | no racial predilection | congenital reticular hyperpigmentation, especially on the neck and chest, with leukoplakia and nail atrophy in fingernails and toenails | hematologic abnormalities |
| Naegeli–Franceschetti–Jadassohn syndrome | KRT14 | no racial predilection | reticulate hyperpigmentation on the neck, chest, abdomen, and axillae | hypoplasia of dermatoglyphics, dental anomalies, diffuse thickening of the palms and feet, hypohidrosis, nail dystrophy |
| Dermatopathia pigmentosa reticularis | KRT14 | no racial predilection | reticulate hyperpigmentation located primarily on the trunk | aplasia of dermatoglyphics, noncicatricial alopecia, hypohidrosis, nail dystrophy |
| X-linked reticulate pigmentary disorder | POLA1 | no racial predilection | male: reticulate hyperpigmentation and hypopigmentation; female: patchy pigmentation along the lines of Blaschko | male: upswept frontal hairline, flared eyebrows, hypohidrosis, gastrointestinal inflammation, recurrent respiratory infections, failure to thrive; female: lack of systemic manifestations |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, M.-H.; Chou, P.-C.; Lee, I.-C.; Yang, S.-F.; Yu, H.-S.; Yu, S. Inherited Reticulate Pigmentary Disorders. Genes 2023, 14, 1300. https://doi.org/10.3390/genes14061300
Lin M-H, Chou P-C, Lee I-C, Yang S-F, Yu H-S, Yu S. Inherited Reticulate Pigmentary Disorders. Genes. 2023; 14(6):1300. https://doi.org/10.3390/genes14061300
Chicago/Turabian StyleLin, Min-Huei, Pei-Chen Chou, I-Chen Lee, Syuan-Fei Yang, Hsin-Su Yu, and Sebastian Yu. 2023. "Inherited Reticulate Pigmentary Disorders" Genes 14, no. 6: 1300. https://doi.org/10.3390/genes14061300
APA StyleLin, M.-H., Chou, P.-C., Lee, I.-C., Yang, S.-F., Yu, H.-S., & Yu, S. (2023). Inherited Reticulate Pigmentary Disorders. Genes, 14(6), 1300. https://doi.org/10.3390/genes14061300