
Hypertrophic Cardiomyopathy Complicated by Post-COVID-19 Myopericarditis in Patient with ANO5-Related Distal Myopathy
 , ,
, ,  , , and
, , and {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
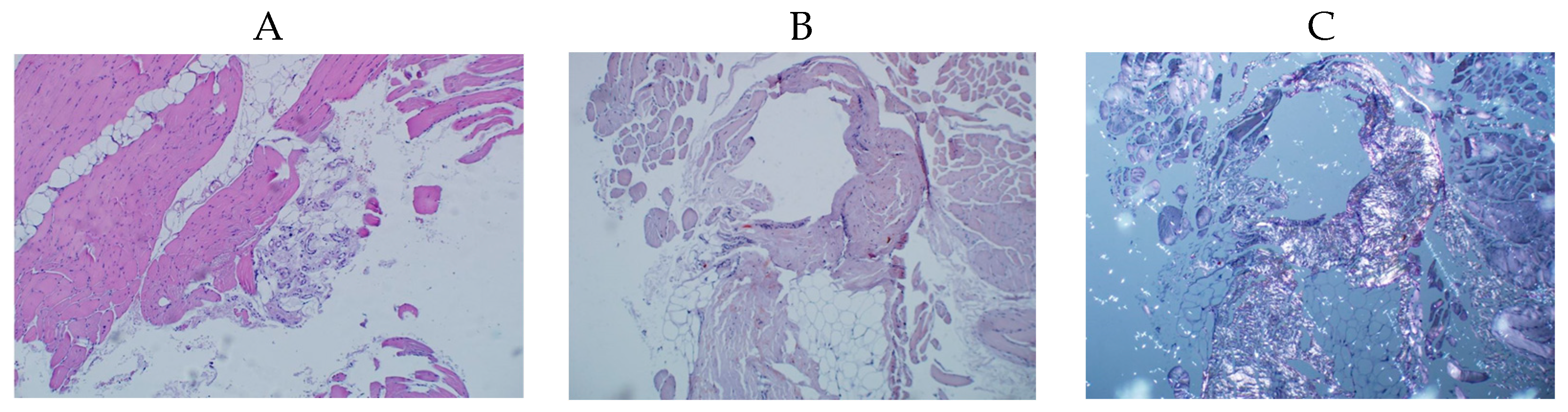
3.1. Case Presentation
3.2. Differential Diagnostics and Treatment Strategy
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cannata, A.; Artico, J.; Gentile, P.; Merlo, M.; Sinagra, G. Myocarditis evolving in cardiomyopathy: When genetics and offending causes work together. Eur. Heart J. Suppl. 2019, 21, B90–B95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campuzano, O.; Fernández-Falgueras, A.; Sarquella-Brugada, G.; Sanchez, O.; Cesar, S.; Mademont, I.; Allegue, C.; Mates, J.; Pérez-Serra, A.; Coll, M.; et al. Genetically Vulnerable Myocardium May Predispose to Myocarditis. J. Am. Coll. Cardiol. 2015, 66, 2913–2914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbustini, E.; Narula, N.; Giuliani, L.; Di Toro, A. Genetic Basis of Myocarditis: Myth or Reality? Myocarditis. Myocarditis 2020, 7, 45–89. [Google Scholar]
- Frustaci, A.; Verardo, R.; Caldarulo, M.; Acconcia, M.C.; Russo, M.A.; Chimenti, C. Myocarditis in hypertrophic cardiomyopathy patients presenting acute clinical deterioration. Eur. Heart J. 2007, 28, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Basso, C. Myocarditis in hypertrophic cardiomyopathy. Eur. Heart J. 2007, 28, 1663–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christiansen, J.; Güttsches, A.K.; Schara-Schmidt, U.; Vorgerd, M.; Heute, C.; Preusse, C.; Stenzel, W.; Roos, A. ANO5-related muscle diseases: From clinics and genetics to pathology and research strategies. Genes Dis. 2022, 9, 1506–1520. [Google Scholar] [CrossRef] [PubMed]
- Wahbi, K.; Béhin, A.; Bécane, H.M.; Leturcq, F.; Cossée, M.; Laforêt, P.; Stojkovic, T.; Carlier, P.; Toussaint, M.; Gaxotte, V.; et al. Dilated cardiomyopathy in patients with mutations in anoctamin 5. Int. J. Cardiol. 2013, 168, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Soontrapa, P.; Liewluck, T. Anoctamin 5 (ANO5) Muscle Disorders: A Narrative Review. Genes 2022, 13, 1736. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, A.M.S.; Coimbra-Neto, A.R.; Souza, P.V.S.; Winckler, P.B.; Gonçalves, M.V.M.; Cavalcanti, E.B.U.; Carvalho, A.A.D.S.; Sobreira, C.F.D.R.; Camelo, C.G.; Mendonça, R.D.H.; et al. Clinical and molecular findings in a cohort of ANO5-related myopathy. Ann. Clin. Transl. Neurol. 2019, 6, 1225–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penttilä, S.; Palmio, J.; Suominen, T.; Raheem, O.; Evilä, A.; Muelas Gomez, N.; Tasca, G.; Waddell, L.B.; Clarke, N.F.; Barboi, A.; et al. Eight new mutations and the expanding phenotype variability in muscular dystrophy caused by ANO5. Neurology 2012, 78, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Anandan, C.; Milone, M.; Liewluck, T. Intramuscular interstitial amyloid deposition does not impact anoctaminopathy-5 phenotype. Muscle Nerve 2019, 59, 133–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Kooi, A.J.; Ten Dam, L.; Frankhuizen, W.S.; Straathof, C.S.; van Doorn, P.A.; de Visser, M.; Ginjaar, I.B. ANO5 mutations in the Dutch limb girdle muscular dystrophy population. Neuromuscul. Disord. 2013, 23, 456–460. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Desai, M.Y.; Nishimura, R.A.; Spirito, P.; Rakowski, H.; Towbin, J.A.; Rowin, E.J.; Maron, M.S.; Sherrid, M.V. Diagnosis and Evaluation of Hypertrophic Cardiomyopathy: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2022, 79, 372–389. [Google Scholar] [CrossRef] [PubMed]
- Blagova, O.; Lutokhina, Y.; Kogan, E.; Kukleva, A.; Ainetdinova, D.; Novosadov, V.; Rud`, R.; Savina, P.; Zaitsev, A.; Fomin, V. Chronic biopsy proven post-COVID myoendocarditis with SARS-Cov-2 persistence and high level of antiheart antibodies. Clin. Cardiol. 2022, 45, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Vojdani, A.; Kharrazian, D. Potential antigenic cross- reactivitybetween SARS- CoV-2 and human tissue with a possible link to an increase in autoimmune diseases. Clin. Immunol. 2020, 217, 108480. [Google Scholar] [CrossRef] [PubMed]
- Del Valle, D.M.; Kim-Schulze, S.; Huang, H.H.; Beckmann, N.D.; Nirenberg, S.; Wang, B.; Lavin, Y.; Swartz, T.H.; Madduri, D.; Stock, A.; et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020, 26, 1636–1643. [Google Scholar] [CrossRef] [PubMed]
- Hanna, A.; Frangogiannis, N.G. Inflammatory Cytokines and Chemokines as Therapeutic Targets in Heart Failure. Cardiovasc. Drugs Ther. 2020, 34, 849–863. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blagova, O.; Lutokhina, Y.; Vukolova, M.; Pirozhkov, S.; Sarkisova, N.; Ainetdinova, D.; Das, A.; Krot, M.; Smolyannikova, V.; Litvitsky, P.; et al. Hypertrophic Cardiomyopathy Complicated by Post-COVID-19 Myopericarditis in Patient with ANO5-Related Distal Myopathy. Genes 2023, 14, 1332. https://doi.org/10.3390/genes14071332
Blagova O, Lutokhina Y, Vukolova M, Pirozhkov S, Sarkisova N, Ainetdinova D, Das A, Krot M, Smolyannikova V, Litvitsky P, et al. Hypertrophic Cardiomyopathy Complicated by Post-COVID-19 Myopericarditis in Patient with ANO5-Related Distal Myopathy. Genes. 2023; 14(7):1332. https://doi.org/10.3390/genes14071332
Chicago/Turabian StyleBlagova, Olga, Yulia Lutokhina, Marina Vukolova, Sergey Pirozhkov, Natalia Sarkisova, Dilara Ainetdinova, Anushree Das, Marina Krot, Vera Smolyannikova, Petr Litvitsky, and et al. 2023. "Hypertrophic Cardiomyopathy Complicated by Post-COVID-19 Myopericarditis in Patient with ANO5-Related Distal Myopathy" Genes 14, no. 7: 1332. https://doi.org/10.3390/genes14071332